We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
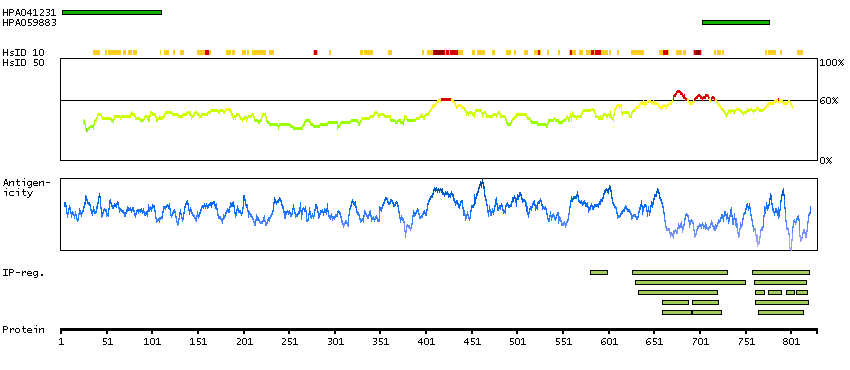
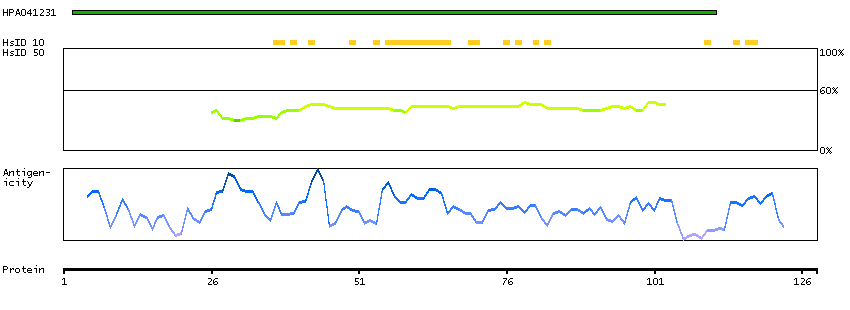
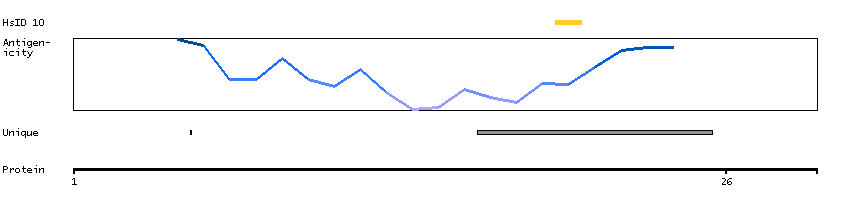
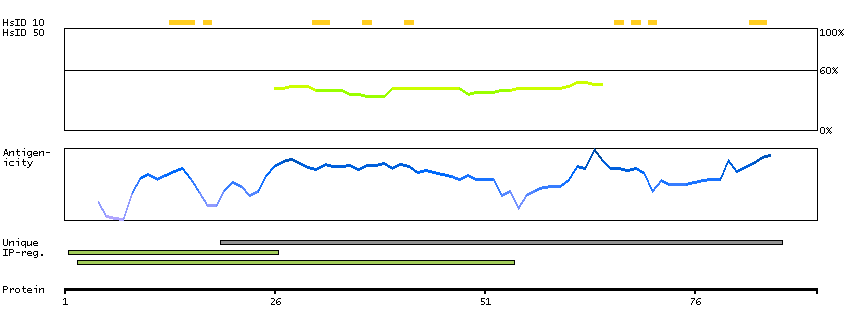
Protein phosphatase 1, regulatory subunit 13 like (HGNC Symbol)
Entrez gene summary
IASPP is one of the most evolutionarily conserved inhibitors of p53 (TP53; MIM 191170), whereas ASPP1 (MIM 606455) and ASPP2 (MIM 602143) are activators of p53.[supplied by OMIM, Mar 2008]