We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
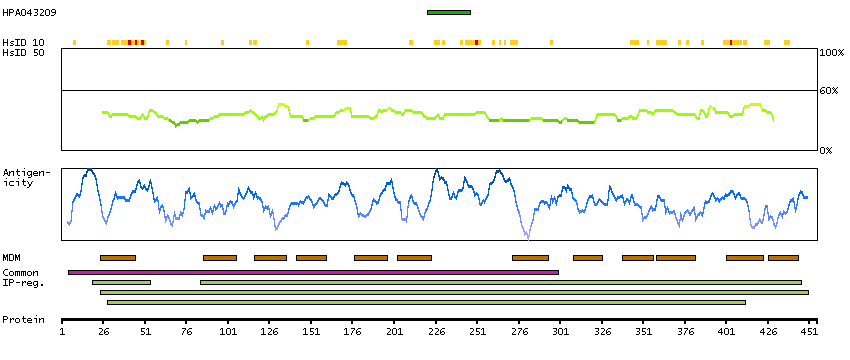
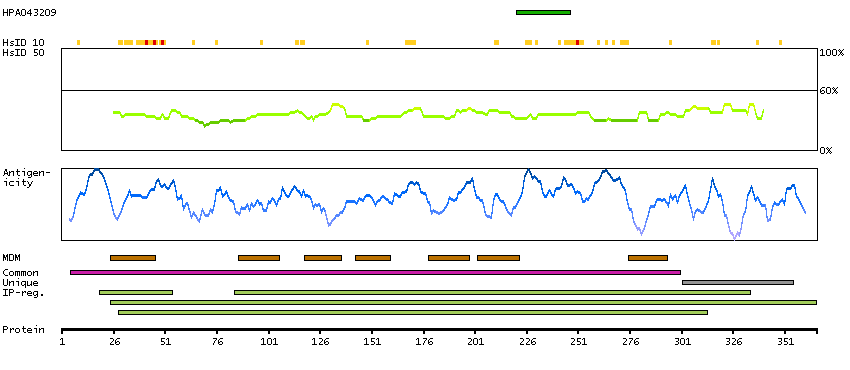
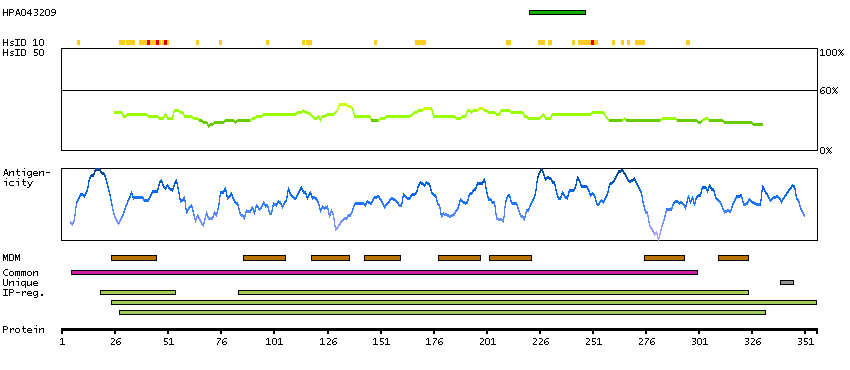
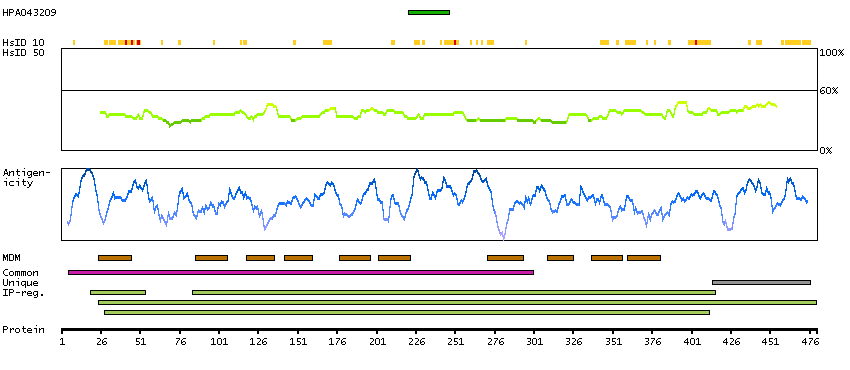
Major facilitator superfamily domain containing 10 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the major facilitator superfamily of transporter proteins. The encoded protein likely functions in efflux of organic anions, including the non-steroidal anti-inflammatory drugs indomethacin and diclofenac. Alternatively spliced transcript variants have been described. [provided by RefSeq, Mar 2009]