We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
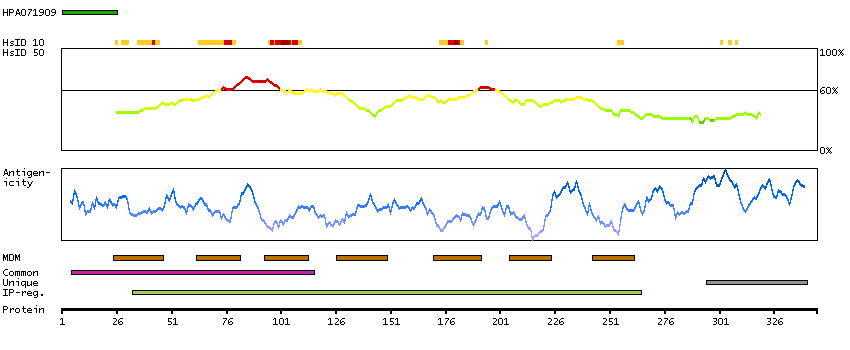
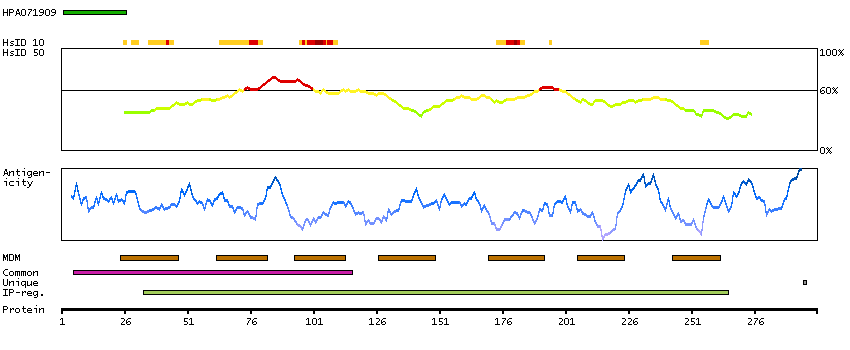
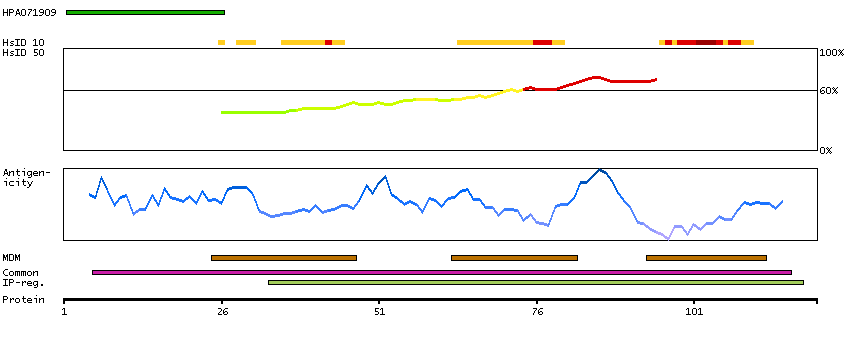
G protein-coupled receptor, class C, group 5, member D (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene is a member of the G protein-coupled receptor family; however, the specific function of this gene has not yet been determined. [provided by RefSeq, Jul 2008]