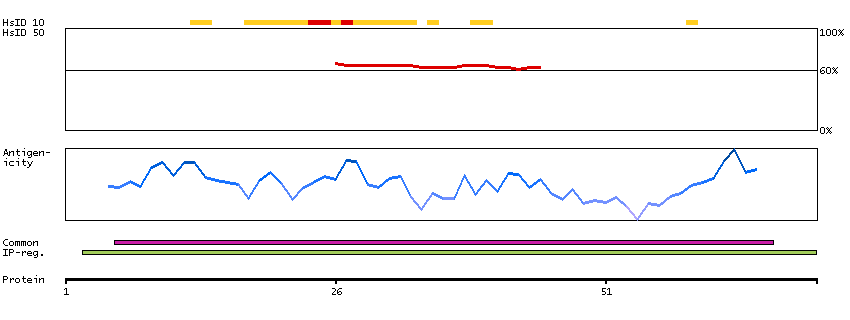
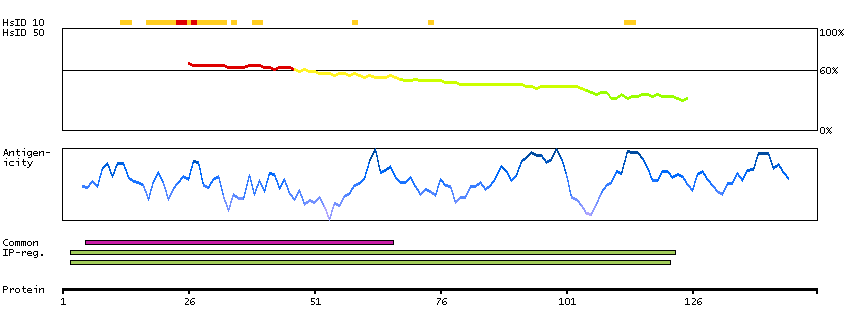
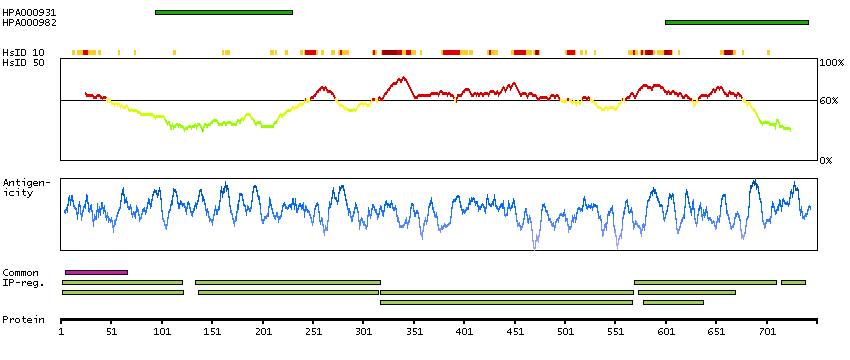
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Signal transducer and activator of transcription 1, 91kDa (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene is a member of the STAT protein family. In response to cytokines and growth factors, STAT family members are phosphorylated by the receptor associated kinases, and then form homo- or heterodimers that translocate to the cell nucleus where they act as transcription activators. This protein can be activated by various ligands including interferon-alpha, interferon-gamma, EGF, PDGF and IL6. This protein mediates the expression of a variety of genes, which is thought to be important for cell viability in response to different cell stimuli and pathogens. Two alternatively spliced transcript variants encoding distinct isoforms have been described. [provided by RefSeq, Jul 2008]
P42224 [Direct mapping] Signal transducer and activator of transcription 1-alpha/beta
Show all
Predicted intracellular proteins Plasma proteins Transcription factors Immunoglobulin fold Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000790 [nuclear chromatin] GO:0000978 [RNA polymerase II core promoter proximal region sequence-specific DNA binding] GO:0000979 [RNA polymerase II core promoter sequence-specific DNA binding] GO:0000983 [transcription factor activity, RNA polymerase II core promoter sequence-specific] GO:0001937 [negative regulation of endothelial cell proliferation] GO:0002053 [positive regulation of mesenchymal cell proliferation] GO:0003340 [negative regulation of mesenchymal to epithelial transition involved in metanephros morphogenesis] GO:0003690 [double-stranded DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0004871 [signal transducer activity] GO:0005164 [tumor necrosis factor receptor binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0005829 [cytosol] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0006915 [apoptotic process] GO:0007165 [signal transduction] GO:0007259 [JAK-STAT cascade] GO:0008015 [blood circulation] GO:0009617 [response to bacterium] GO:0016032 [viral process] GO:0016525 [negative regulation of angiogenesis] GO:0019221 [cytokine-mediated signaling pathway] GO:0019899 [enzyme binding] GO:0030424 [axon] GO:0030425 [dendrite] GO:0031663 [lipopolysaccharide-mediated signaling pathway] GO:0032496 [response to lipopolysaccharide] GO:0033209 [tumor necrosis factor-mediated signaling pathway] GO:0034097 [response to cytokine] GO:0034240 [negative regulation of macrophage fusion] GO:0035456 [response to interferon-beta] GO:0035458 [cellular response to interferon-beta] GO:0042802 [identical protein binding] GO:0042803 [protein homodimerization activity] GO:0042981 [regulation of apoptotic process] GO:0043124 [negative regulation of I-kappaB kinase/NF-kappaB signaling] GO:0043330 [response to exogenous dsRNA] GO:0043434 [response to peptide hormone] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046725 [negative regulation by virus of viral protein levels in host cell] GO:0048471 [perinuclear region of cytoplasm] GO:0048661 [positive regulation of smooth muscle cell proliferation] GO:0051591 [response to cAMP] GO:0051607 [defense response to virus] GO:0060333 [interferon-gamma-mediated signaling pathway] GO:0060334 [regulation of interferon-gamma-mediated signaling pathway] GO:0060337 [type I interferon signaling pathway] GO:0060338 [regulation of type I interferon-mediated signaling pathway] GO:0060397 [JAK-STAT cascade involved in growth hormone signaling pathway] GO:0061326 [renal tubule development] GO:0071222 [cellular response to lipopolysaccharide] GO:0071345 [cellular response to cytokine stimulus] GO:0071407 [cellular response to organic cyclic compound] GO:0072136 [metanephric mesenchymal cell proliferation involved in metanephros development] GO:0072162 [metanephric mesenchymal cell differentiation] GO:0072308 [negative regulation of metanephric nephron tubule epithelial cell differentiation]
P42224 [Direct mapping] Signal transducer and activator of transcription 1-alpha/beta
Show all
Predicted intracellular proteins Plasma proteins Transcription factors Immunoglobulin fold Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000790 [nuclear chromatin] GO:0000978 [RNA polymerase II core promoter proximal region sequence-specific DNA binding] GO:0000979 [RNA polymerase II core promoter sequence-specific DNA binding] GO:0000983 [transcription factor activity, RNA polymerase II core promoter sequence-specific] GO:0001937 [negative regulation of endothelial cell proliferation] GO:0002053 [positive regulation of mesenchymal cell proliferation] GO:0003340 [negative regulation of mesenchymal to epithelial transition involved in metanephros morphogenesis] GO:0003690 [double-stranded DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0004871 [signal transducer activity] GO:0005164 [tumor necrosis factor receptor binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0005829 [cytosol] GO:0006355 [regulation of transcription, DNA-templated] GO:0006915 [apoptotic process] GO:0007165 [signal transduction] GO:0007259 [JAK-STAT cascade] GO:0008015 [blood circulation] GO:0016032 [viral process] GO:0016525 [negative regulation of angiogenesis] GO:0019221 [cytokine-mediated signaling pathway] GO:0019899 [enzyme binding] GO:0030424 [axon] GO:0030425 [dendrite] GO:0033209 [tumor necrosis factor-mediated signaling pathway] GO:0034097 [response to cytokine] GO:0035456 [response to interferon-beta] GO:0035458 [cellular response to interferon-beta] GO:0042802 [identical protein binding] GO:0042803 [protein homodimerization activity] GO:0042981 [regulation of apoptotic process] GO:0043124 [negative regulation of I-kappaB kinase/NF-kappaB signaling] GO:0043434 [response to peptide hormone] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046725 [negative regulation by virus of viral protein levels in host cell] GO:0048471 [perinuclear region of cytoplasm] GO:0048661 [positive regulation of smooth muscle cell proliferation] GO:0051591 [response to cAMP] GO:0051607 [defense response to virus] GO:0060333 [interferon-gamma-mediated signaling pathway] GO:0060334 [regulation of interferon-gamma-mediated signaling pathway] GO:0060337 [type I interferon signaling pathway] GO:0060338 [regulation of type I interferon-mediated signaling pathway] GO:0060397 [JAK-STAT cascade involved in growth hormone signaling pathway] GO:0061326 [renal tubule development] GO:0072136 [metanephric mesenchymal cell proliferation involved in metanephros development] GO:0072162 [metanephric mesenchymal cell differentiation] GO:0072308 [negative regulation of metanephric nephron tubule epithelial cell differentiation]
P42224 [Direct mapping] Signal transducer and activator of transcription 1-alpha/beta
Show all
Predicted intracellular proteins Plasma proteins Transcription factors Immunoglobulin fold Disease related genes Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000790 [nuclear chromatin] GO:0000978 [RNA polymerase II core promoter proximal region sequence-specific DNA binding] GO:0000979 [RNA polymerase II core promoter sequence-specific DNA binding] GO:0000983 [transcription factor activity, RNA polymerase II core promoter sequence-specific] GO:0001937 [negative regulation of endothelial cell proliferation] GO:0002053 [positive regulation of mesenchymal cell proliferation] GO:0003340 [negative regulation of mesenchymal to epithelial transition involved in metanephros morphogenesis] GO:0003690 [double-stranded DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0004871 [signal transducer activity] GO:0005164 [tumor necrosis factor receptor binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0005829 [cytosol] GO:0006355 [regulation of transcription, DNA-templated] GO:0006915 [apoptotic process] GO:0007165 [signal transduction] GO:0007259 [JAK-STAT cascade] GO:0008015 [blood circulation] GO:0016032 [viral process] GO:0016525 [negative regulation of angiogenesis] GO:0019221 [cytokine-mediated signaling pathway] GO:0019899 [enzyme binding] GO:0030424 [axon] GO:0030425 [dendrite] GO:0033209 [tumor necrosis factor-mediated signaling pathway] GO:0034097 [response to cytokine] GO:0035456 [response to interferon-beta] GO:0035458 [cellular response to interferon-beta] GO:0042802 [identical protein binding] GO:0042803 [protein homodimerization activity] GO:0042981 [regulation of apoptotic process] GO:0043124 [negative regulation of I-kappaB kinase/NF-kappaB signaling] GO:0043434 [response to peptide hormone] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046725 [negative regulation by virus of viral protein levels in host cell] GO:0048471 [perinuclear region of cytoplasm] GO:0048661 [positive regulation of smooth muscle cell proliferation] GO:0051591 [response to cAMP] GO:0051607 [defense response to virus] GO:0060333 [interferon-gamma-mediated signaling pathway] GO:0060334 [regulation of interferon-gamma-mediated signaling pathway] GO:0060337 [type I interferon signaling pathway] GO:0060338 [regulation of type I interferon-mediated signaling pathway] GO:0060397 [JAK-STAT cascade involved in growth hormone signaling pathway] GO:0061326 [renal tubule development] GO:0072136 [metanephric mesenchymal cell proliferation involved in metanephros development] GO:0072162 [metanephric mesenchymal cell differentiation] GO:0072308 [negative regulation of metanephric nephron tubule epithelial cell differentiation]