We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
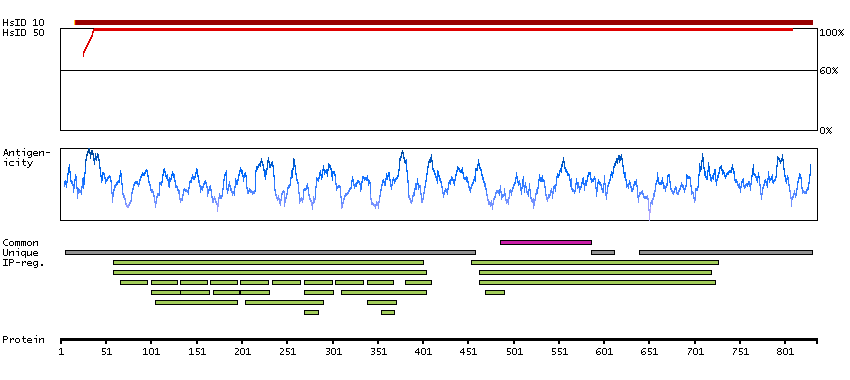
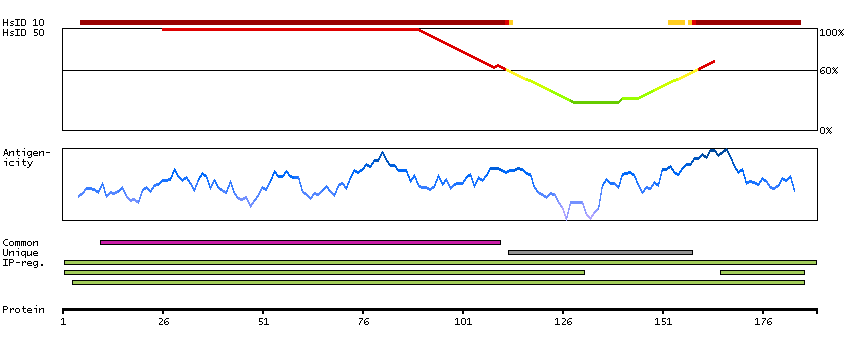
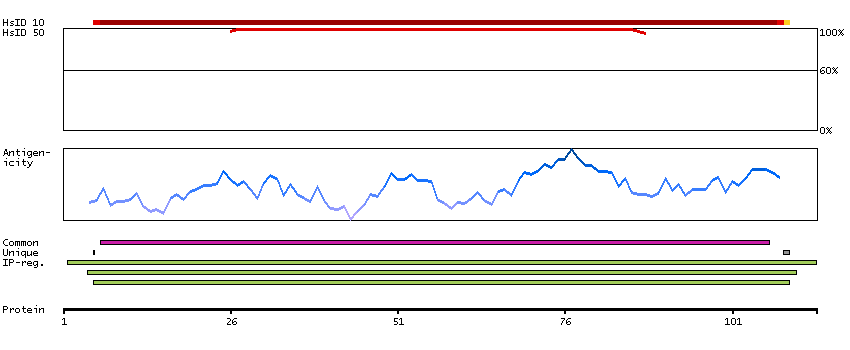
This gene encodes a protein that belongs to the MAP kinase kinase kinase (MAPKKK) family of protein kinases. The protein contains ankyrin repeat, protein kinase and serine-rich domains and is thought to play a role in cardiac physiology. [provided by RefSeq, Sep 2012]