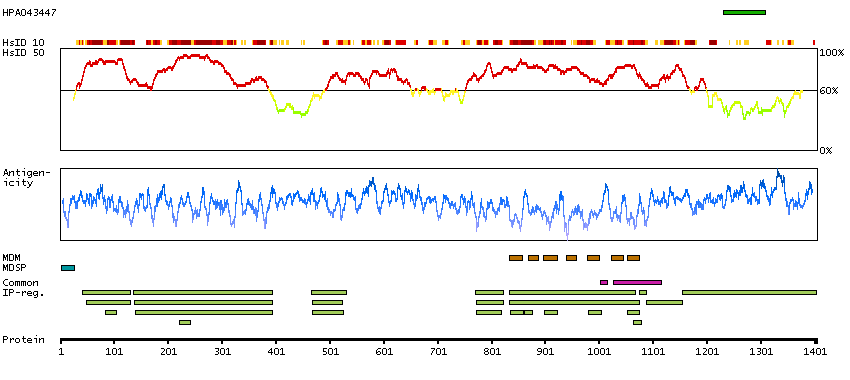
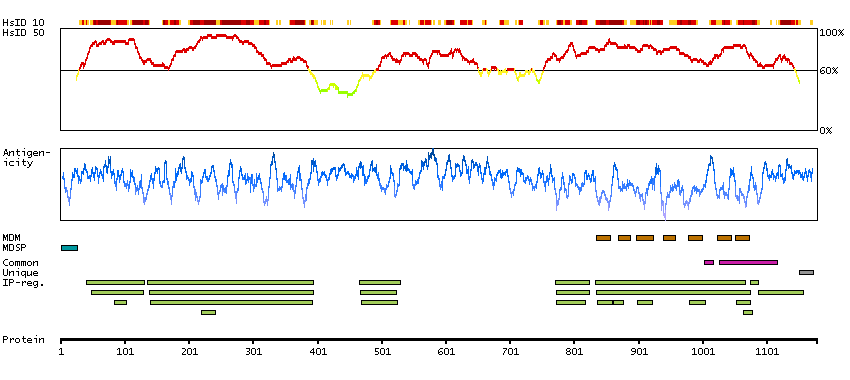
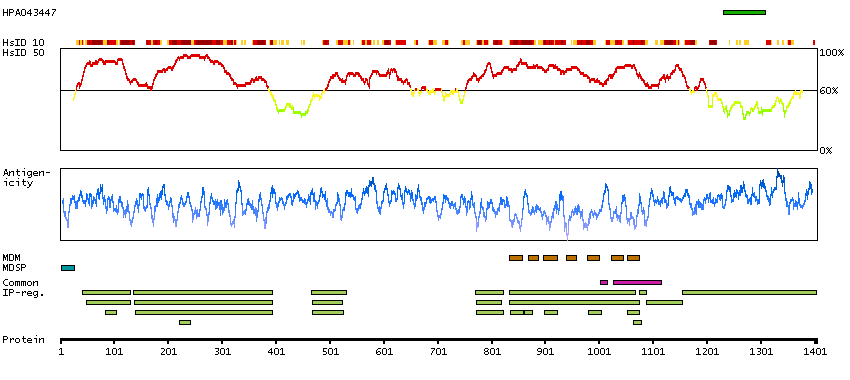
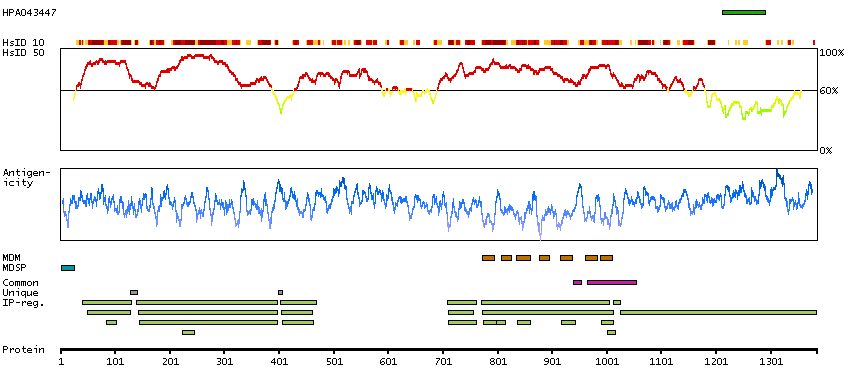
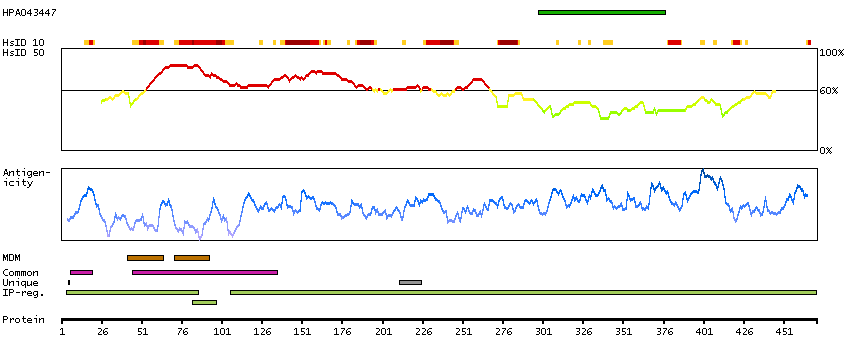
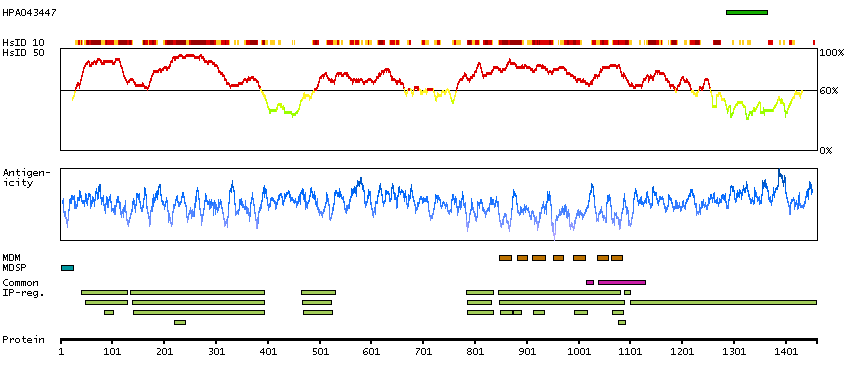
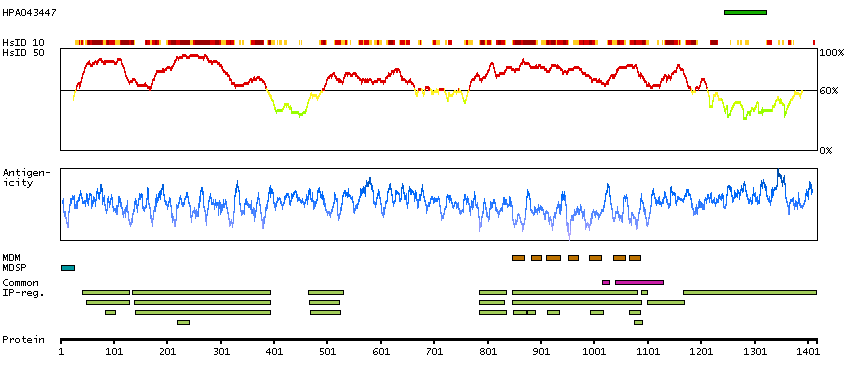
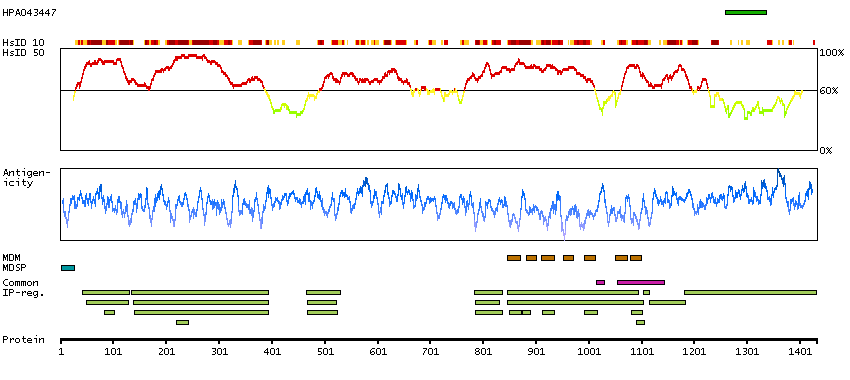
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
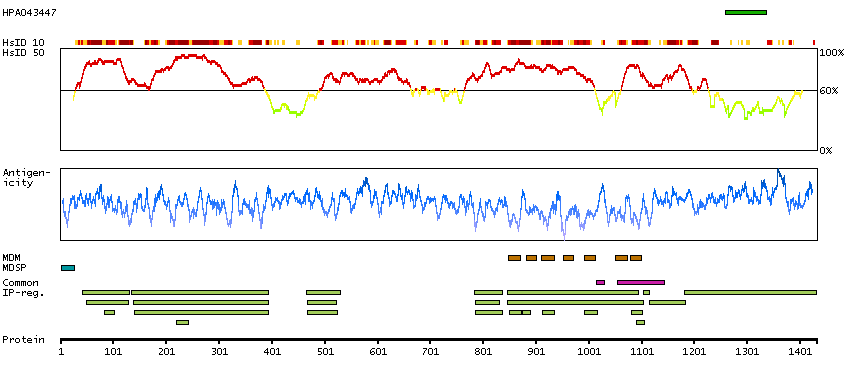
Adhesion G protein-coupled receptor L2 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the latrophilin subfamily of G-protein coupled receptors. The encoded protein participates in the regulation of exocytosis. The proprotein is thought to be further cleaved within a cysteine-rich G-protein-coupled receptor proteolysis site into two chains that are non-covalently bound at the cell membrane. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jul 2014]