We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
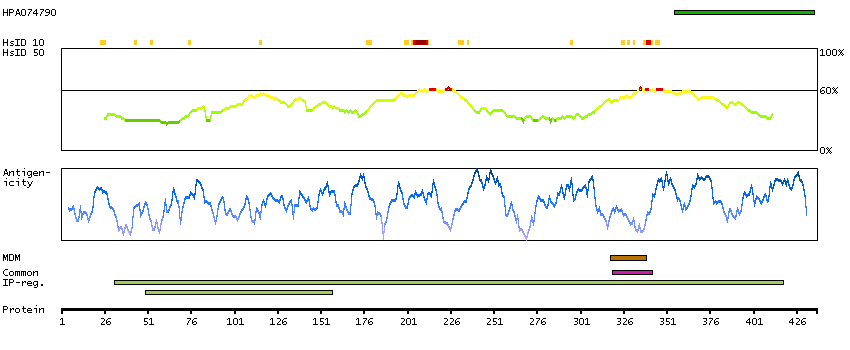
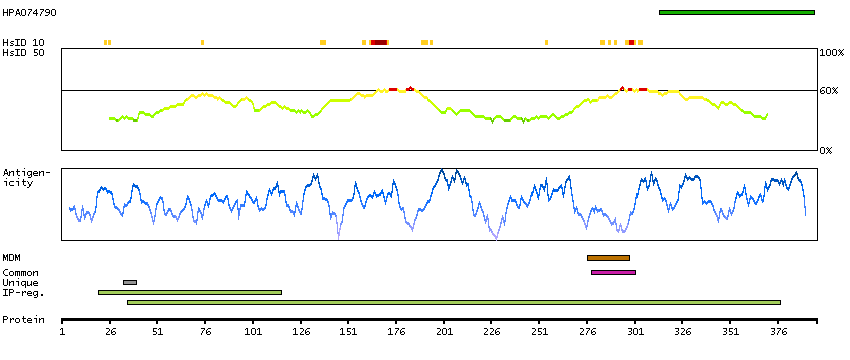
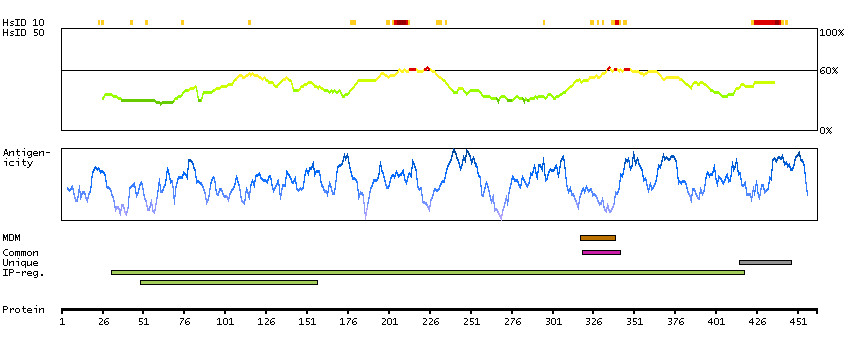
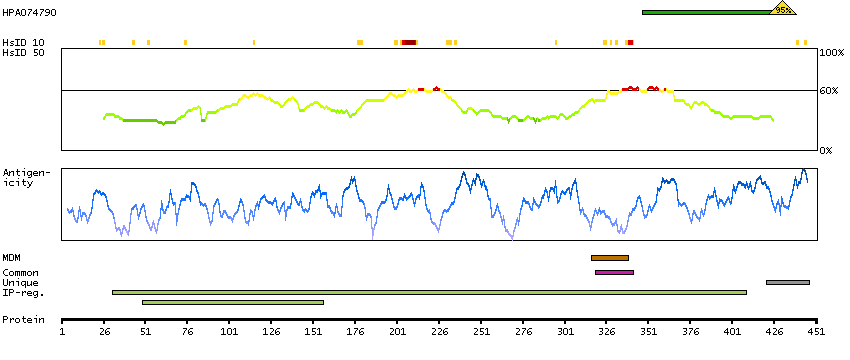
This gene encodes the epsilon member of the sarcoglycan family. Sarcoglycans are transmembrane proteins that are components of the dystrophin-glycoprotein complex, which link the actin cytoskeleton to the extracellular matrix. Unlike other family members which are predominantly expressed in striated muscle, the epsilon sarcoglycan is more broadly expressed. Mutations in this gene are associated with myoclonus-dystonia syndrome. This gene is imprinted, with preferential expression from the paternal allele. Alternatively spliced transcript variants encoding different isoforms have been found for this gene.[provided by RefSeq, Oct 2010]