We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Core-binding factor, runt domain, alpha subunit 2; translocated to, 3 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the myeloid translocation gene family which interact with DNA-bound transcription factors and recruit a range of corepressors to facilitate transcriptional repression. The t(16;21)(q24;q22) translocation is one of the less common karyotypic abnormalities in acute myeloid leukemia. The translocation produces a chimeric gene made up of the 5'-region of the runt-related transcription factor 1 gene fused to the 3'-region of this gene. This gene is also a putative breast tumor suppressor. Alternative splicing results in transcript variants. [provided by RefSeq, Nov 2010]
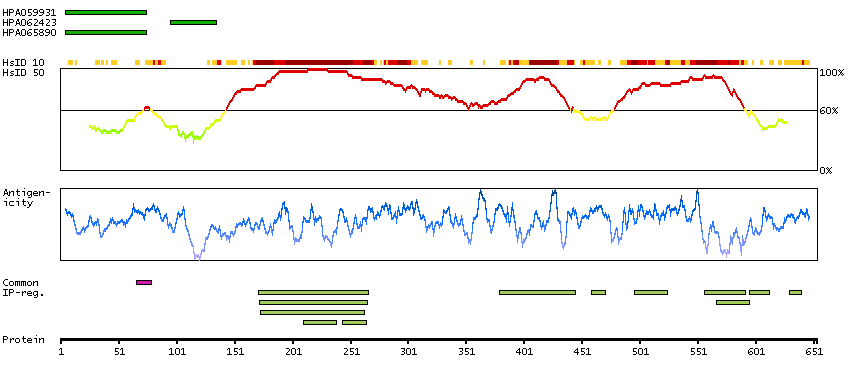
SCAMPI predicted membrane proteins Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Disease related genes Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000139 [Golgi membrane] GO:0001666 [response to hypoxia] GO:0003682 [chromatin binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0003714 [transcription corepressor activity] GO:0005515 [protein binding] GO:0005622 [intracellular] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0008283 [cell proliferation] GO:0008285 [negative regulation of cell proliferation] GO:0030851 [granulocyte differentiation] GO:0032436 [positive regulation of proteasomal ubiquitin-dependent protein catabolic process] GO:0045820 [negative regulation of glycolytic process] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0046872 [metal ion binding] GO:1903715 [regulation of aerobic respiration]
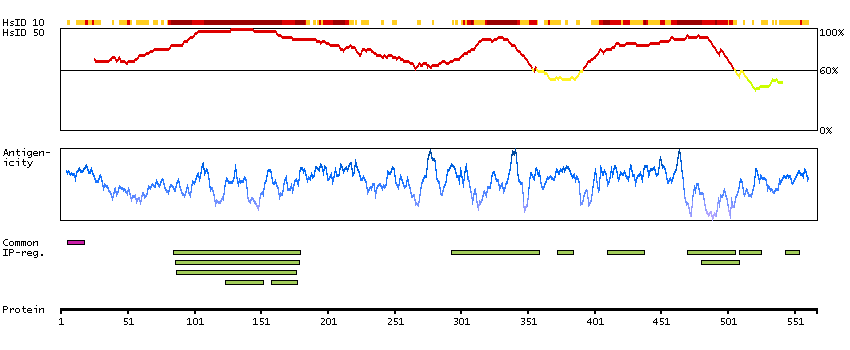
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Disease related genes Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000139 [Golgi membrane] GO:0001666 [response to hypoxia] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0003714 [transcription corepressor activity] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0008283 [cell proliferation] GO:0008285 [negative regulation of cell proliferation] GO:0030851 [granulocyte differentiation] GO:0032436 [positive regulation of proteasomal ubiquitin-dependent protein catabolic process] GO:0045820 [negative regulation of glycolytic process] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0046872 [metal ion binding] GO:1903715 [regulation of aerobic respiration]
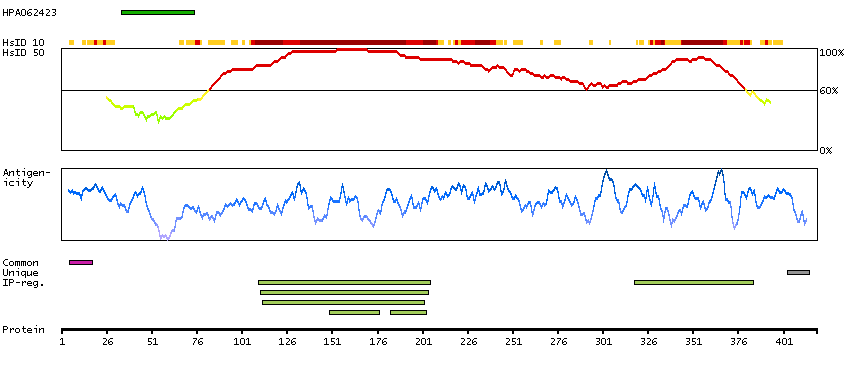
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0003714 [transcription corepressor activity] GO:0005634 [nucleus] GO:0006355 [regulation of transcription, DNA-templated] GO:0045892 [negative regulation of transcription, DNA-templated]
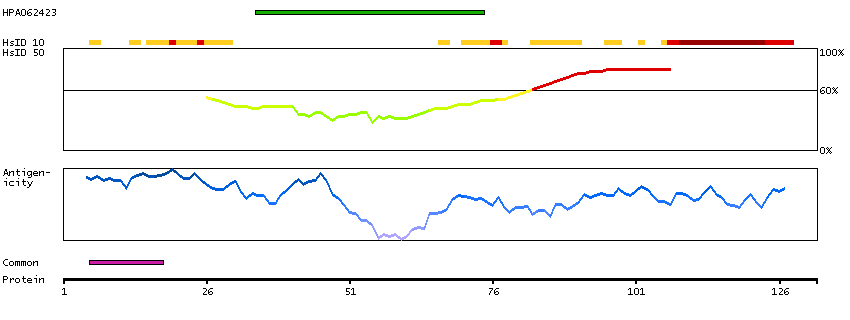
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)