We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
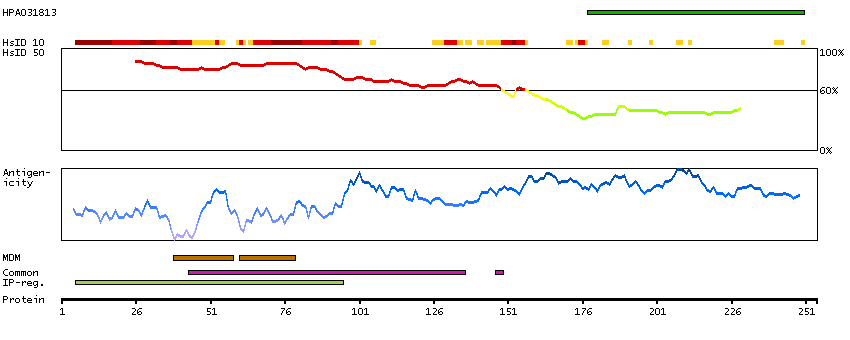
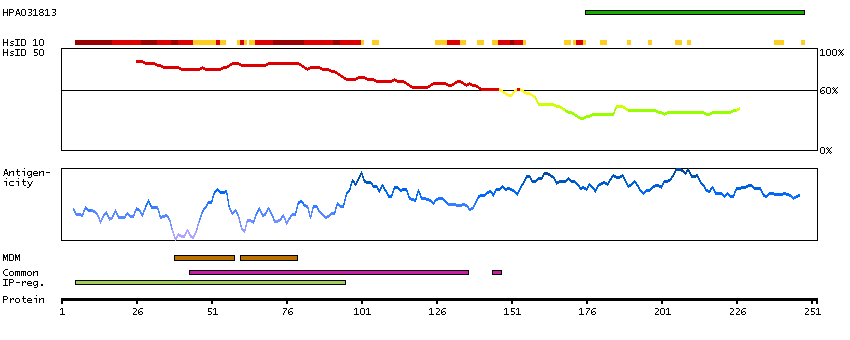
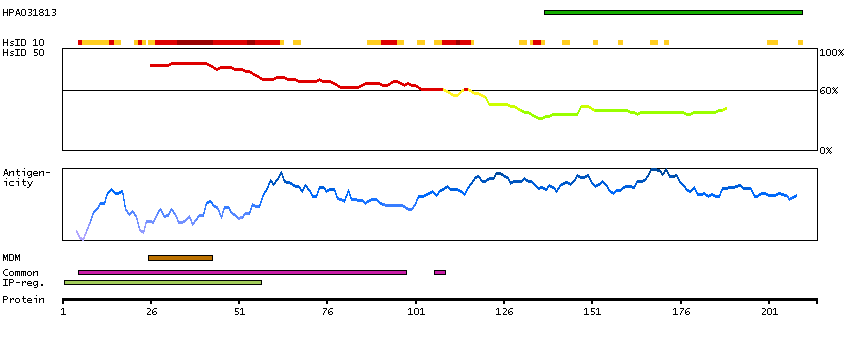
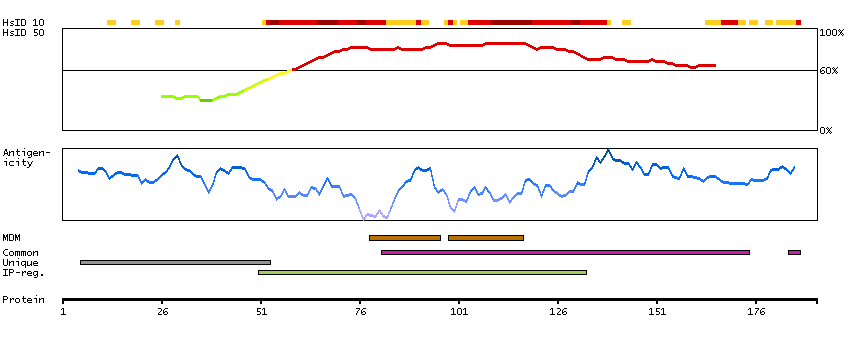
This gene encodes a member of the receptor expression enhancing protein family. Studies of a related gene in mouse suggest that the encoded protein is found in the cell membrane and enhances the function of sweet taste receptors. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Nov 2012]