We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
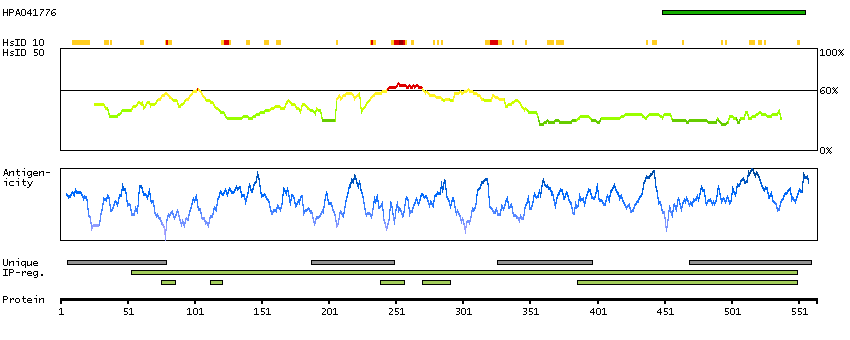
This gene encodes a putative member of the class I family of aminoacyl-tRNA synthetases. These enzymes play a critical role in protein biosynthesis by charging tRNAs with their cognate amino acids. This protein is encoded by the nuclear genome but is likely to be imported to the mitochondrion where it is thought to catalyze the ligation of cysteine to tRNA molecules. A splice-site mutation in this gene has been associated with a novel progressive myoclonic epilepsy disease with similar symptoms to MERRF syndrome. [provided by RefSeq, Mar 2015]
Enzymes ENZYME proteins Ligase SPOCTOPUS predicted secreted proteins Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)