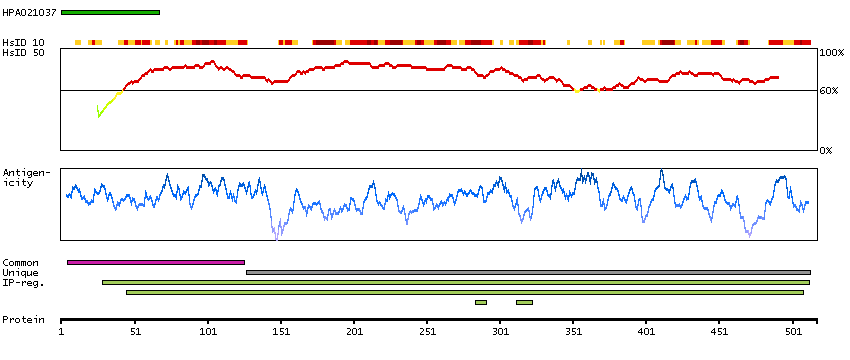
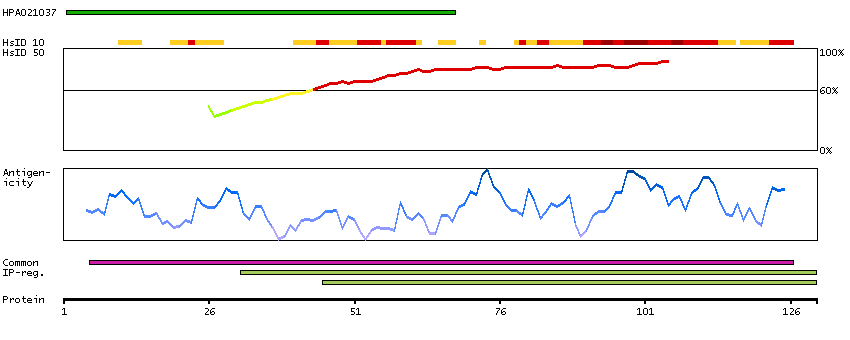
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Aldehyde dehydrogenase 1 family, member B1 (HGNC Symbol)
Entrez gene summary
This protein belongs to the aldehyde dehydrogenases family of proteins. Aldehyde dehydrogenase is the second enzyme of the major oxidative pathway of alcohol metabolism. This gene does not contain introns in the coding sequence. The variation of this locus may affect the development of alcohol-related problems. [provided by RefSeq, Jul 2008]