We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
The protein encoded by this gene belongs to the TDG/mug DNA glycosylase family. Thymine-DNA glycosylase (TDG) removes thymine moieties from G/T mismatches by hydrolyzing the carbon-nitrogen bond between the sugar-phosphate backbone of DNA and the mispaired thymine. With lower activity, this enzyme also removes thymine from C/T and T/T mispairings. TDG can also remove uracil and 5-bromouracil from mispairings with guanine. This enzyme plays a central role in cellular defense against genetic mutation caused by the spontaneous deamination of 5-methylcytosine and cytosine. This gene may have a pseudogene in the p arm of chromosome 12. [provided by RefSeq, Jul 2008]
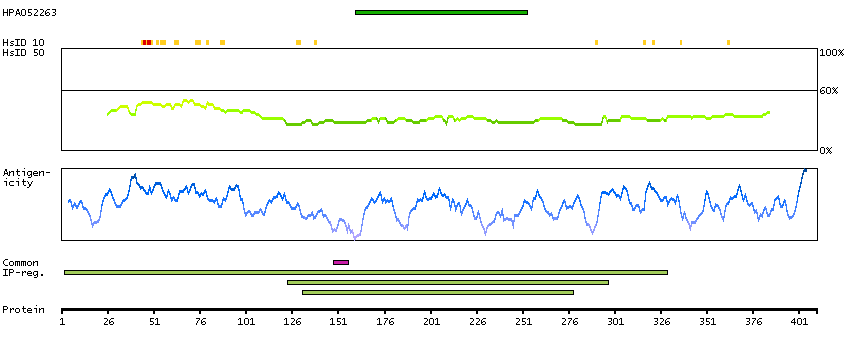
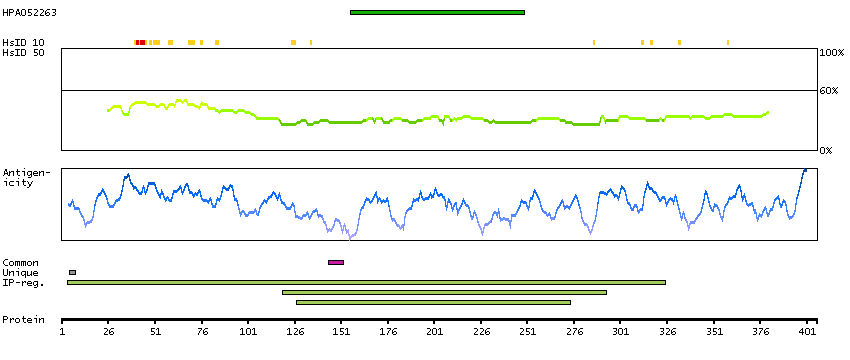
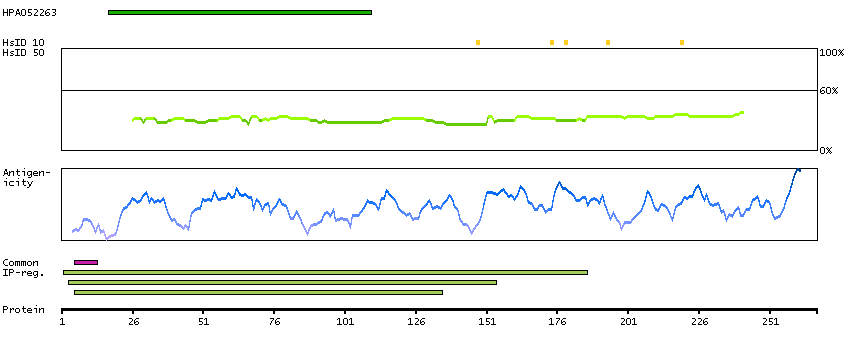
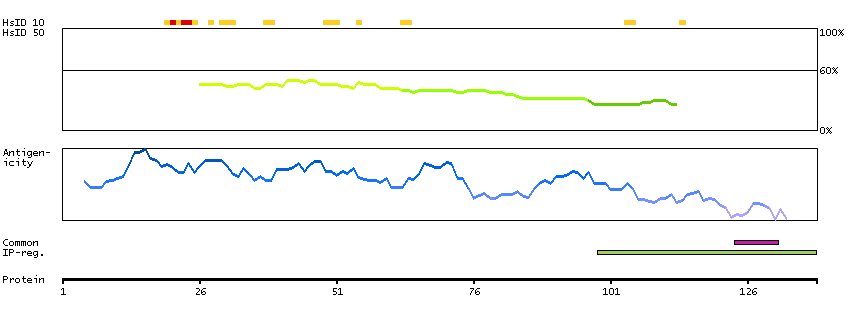
B4E127 [Direct mapping] G/T mismatch-specific thymine DNA glycosylase; cDNA FLJ50939, highly similar to G/T mismatch-specific thymine DNA glycosylase (EC 3.2.2.-)
Show all
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)