We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
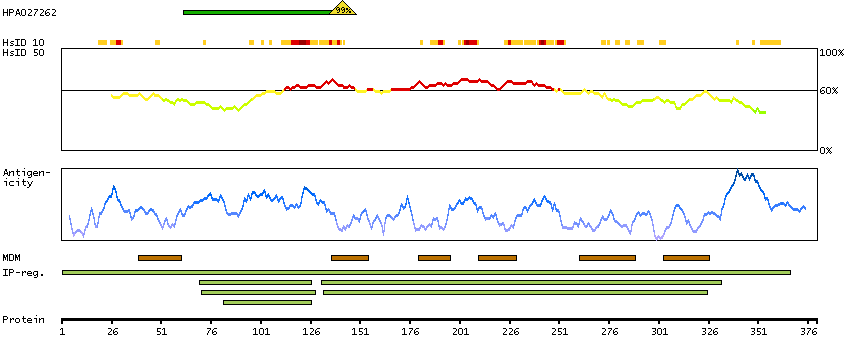
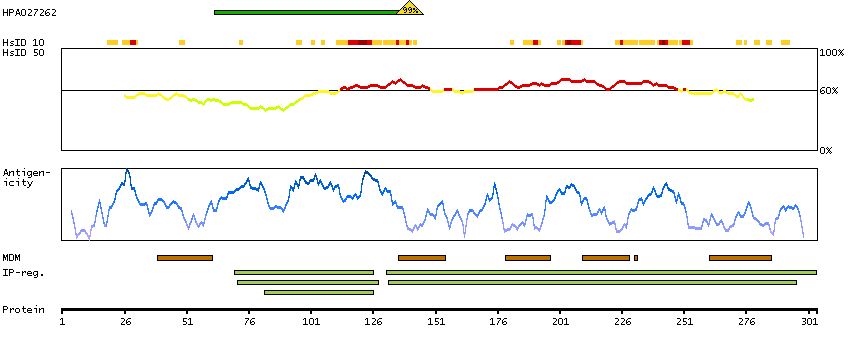
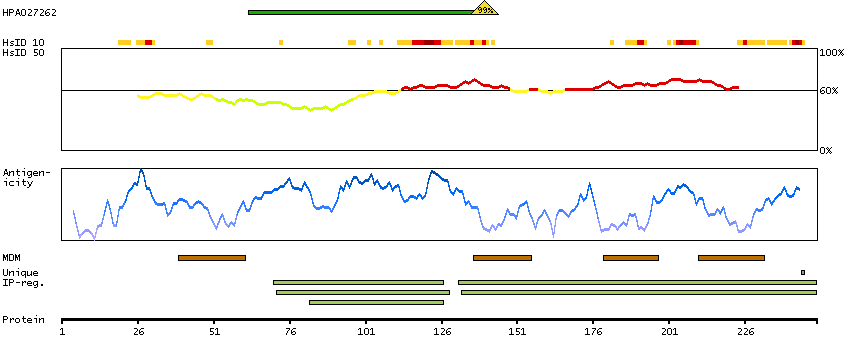
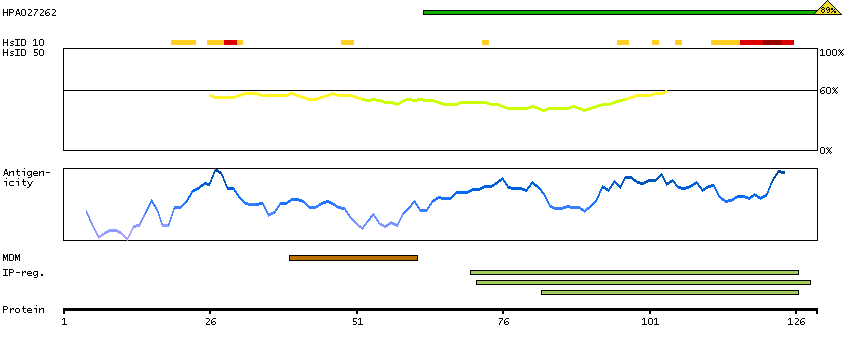
This gene encodes a protein that has sequence similarity to yeast longevity assurance gene 1. Mutation or overexpression of the related gene in yeast has been shown to alter yeast lifespan. The human protein may play a role in the regulation of cell growth. Alternatively spliced transcript variants encoding the same protein have been described. [provided by RefSeq, Jul 2008]