We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
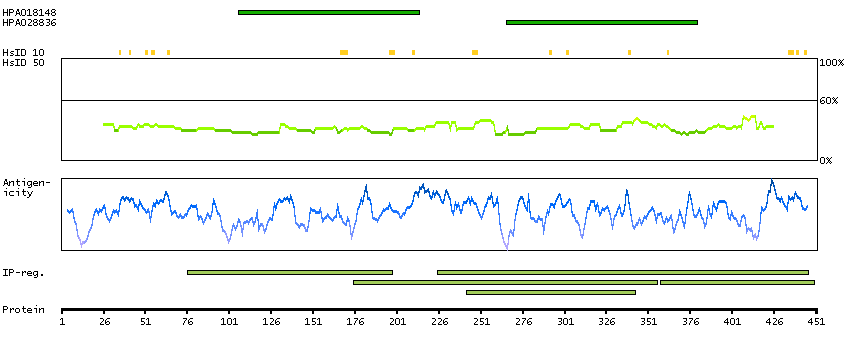
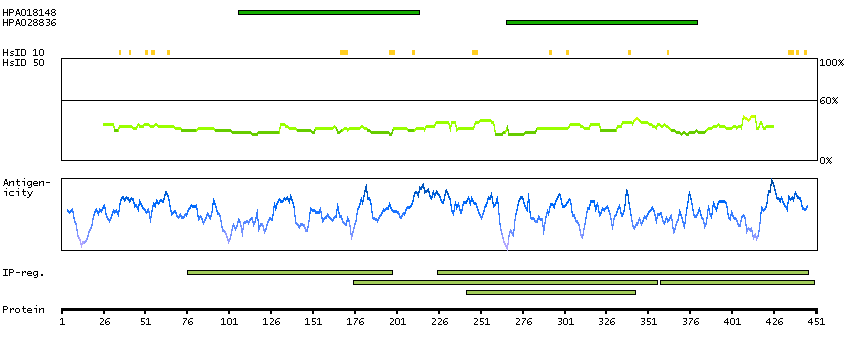
Aminoacyl-tRNA synthetases are a class of enzymes that charge tRNAs with their cognate amino acids. This gene encodes a phenylalanine-tRNA synthetase (PheRS) localized to the mitochondrion which consists of a single polypeptide chain, unlike the (alpha-beta)2 structure of the prokaryotic and eukaryotic cytoplasmic forms of PheRS. Structure analysis and catalytic properties indicate mitochondrial PheRSs may constitute a class of PheRS distinct from the enzymes found in prokaryotes and in the eukaryotic cytoplasm. [provided by RefSeq, Jul 2008]
Enzymes ENZYME proteins Ligase SCAMPI predicted membrane proteins Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Ligase SCAMPI predicted membrane proteins Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Ezkurdia et al 2014)