We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
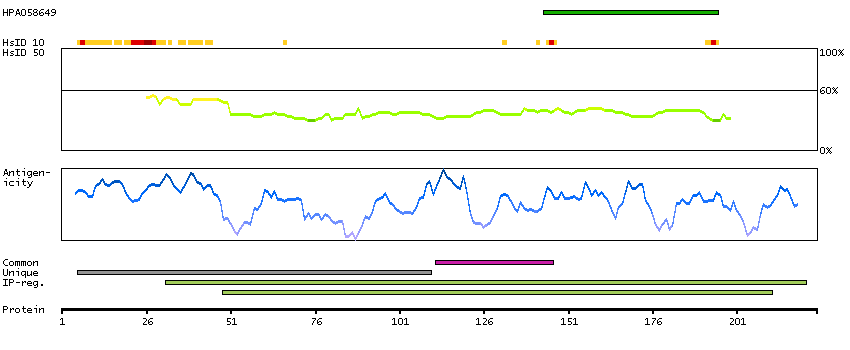
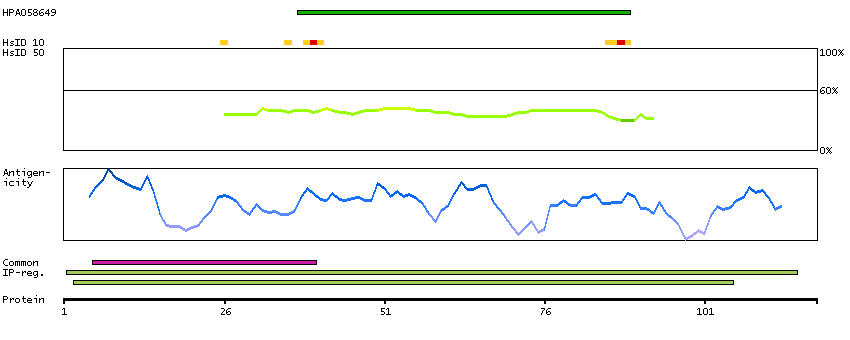
Eukaryotic translation initiation factor 4E family member 3 (HGNC Symbol)
Entrez gene summary
EIF4E3 belongs to the EIF4E family of translational initiation factors that interact with the 5-prime cap structure of mRNA and recruit mRNA to the ribosome (Joshi et al., 2004 [PubMed 15153109]).[supplied by OMIM, Mar 2008]