We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
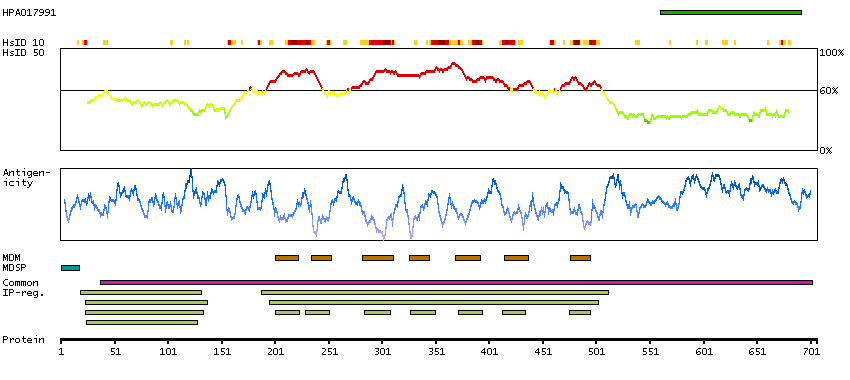
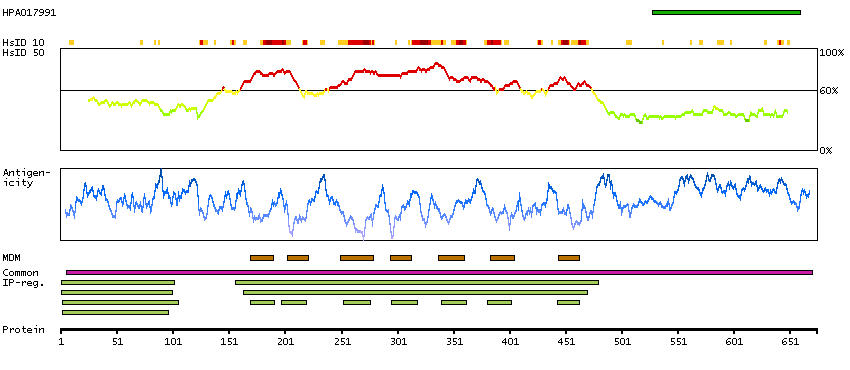
This gene represents a member of the 'frizzled' gene family, which encode 7-transmembrane domain proteins that are receptors for Wnt signaling proteins. The protein encoded by this family member contains a signal peptide, a cysteine-rich domain in the N-terminal extracellular region, and seven transmembrane domains, but unlike other family members, this protein does not contain a C-terminal PDZ domain-binding motif. This protein functions as a negative regulator of the canonical Wnt/beta-catenin signaling cascade, thereby inhibiting the processes that trigger oncogenic transformation, cell proliferation, and inhibition of apoptosis. Alternative splicing results in multiple transcript variants, some of which do not encode a protein with a predicted signal peptide.[provided by RefSeq, Aug 2011]