We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
This gene encodes an integral outer mitochondrial membrane protein that blocks the apoptotic death of some cells such as lymphocytes. Constitutive expression of BCL2, such as in the case of translocation of BCL2 to Ig heavy chain locus, is thought to be the cause of follicular lymphoma. Two transcript variants, produced by alternate splicing, differ in their C-terminal ends. [provided by RefSeq, Jul 2008]
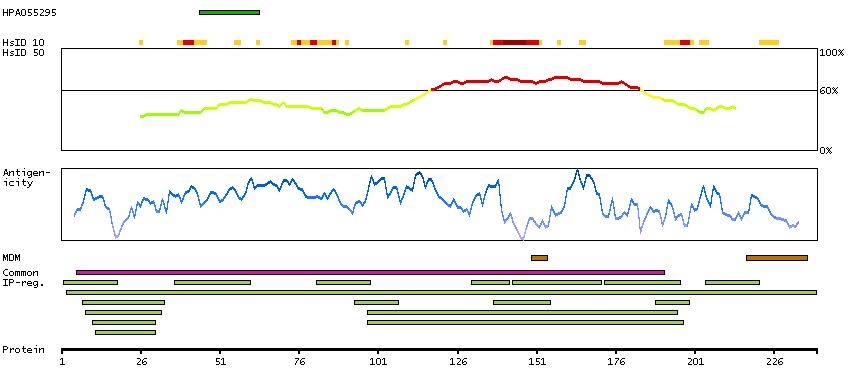
MEMSAT3 predicted membrane proteins SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Plasma proteins Cancer-related genes Candidate cancer biomarkers COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Disease related genes FDA approved drug targets Small molecule drugs Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000209 [protein polyubiquitination] GO:0001836 [release of cytochrome c from mitochondria] GO:0002020 [protease binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005737 [cytoplasm] GO:0005739 [mitochondrion] GO:0005741 [mitochondrial outer membrane] GO:0005783 [endoplasmic reticulum] GO:0005789 [endoplasmic reticulum membrane] GO:0006915 [apoptotic process] GO:0006959 [humoral immune response] GO:0006974 [cellular response to DNA damage stimulus] GO:0007565 [female pregnancy] GO:0008625 [extrinsic apoptotic signaling pathway via death domain receptors] GO:0008630 [intrinsic apoptotic signaling pathway in response to DNA damage] GO:0009314 [response to radiation] GO:0009636 [response to toxic substance] GO:0010039 [response to iron ion] GO:0010507 [negative regulation of autophagy] GO:0012501 [programmed cell death] GO:0015267 [channel activity] GO:0016020 [membrane] GO:0016248 [channel inhibitor activity] GO:0022898 [regulation of transmembrane transporter activity] GO:0030307 [positive regulation of cell growth] GO:0030890 [positive regulation of B cell proliferation] GO:0031625 [ubiquitin protein ligase binding] GO:0031965 [nuclear membrane] GO:0032469 [endoplasmic reticulum calcium ion homeostasis] GO:0032848 [negative regulation of cellular pH reduction] GO:0034097 [response to cytokine] GO:0035094 [response to nicotine] GO:0035872 [nucleotide-binding domain, leucine rich repeat containing receptor signaling pathway] GO:0042100 [B cell proliferation] GO:0042493 [response to drug] GO:0042802 [identical protein binding] GO:0042803 [protein homodimerization activity] GO:0042981 [regulation of apoptotic process] GO:0043066 [negative regulation of apoptotic process] GO:0043496 [regulation of protein homodimerization activity] GO:0043497 [regulation of protein heterodimerization activity] GO:0043524 [negative regulation of neuron apoptotic process] GO:0043565 [sequence-specific DNA binding] GO:0045087 [innate immune response] GO:0046902 [regulation of mitochondrial membrane permeability] GO:0046930 [pore complex] GO:0046982 [protein heterodimerization activity] GO:0050853 [B cell receptor signaling pathway] GO:0051402 [neuron apoptotic process] GO:0051434 [BH3 domain binding] GO:0051607 [defense response to virus] GO:0051881 [regulation of mitochondrial membrane potential] GO:0051902 [negative regulation of mitochondrial depolarization] GO:0051924 [regulation of calcium ion transport] GO:0055085 [transmembrane transport] GO:0070059 [intrinsic apoptotic signaling pathway in response to endoplasmic reticulum stress] GO:0097192 [extrinsic apoptotic signaling pathway in absence of ligand] GO:0097193 [intrinsic apoptotic signaling pathway] GO:1900740 [positive regulation of protein insertion into mitochondrial membrane involved in apoptotic signaling pathway] GO:2000811 [negative regulation of anoikis] GO:2001234 [negative regulation of apoptotic signaling pathway] GO:2001240 [negative regulation of extrinsic apoptotic signaling pathway in absence of ligand] GO:2001243 [negative regulation of intrinsic apoptotic signaling pathway] GO:2001244 [positive regulation of intrinsic apoptotic signaling pathway]