We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
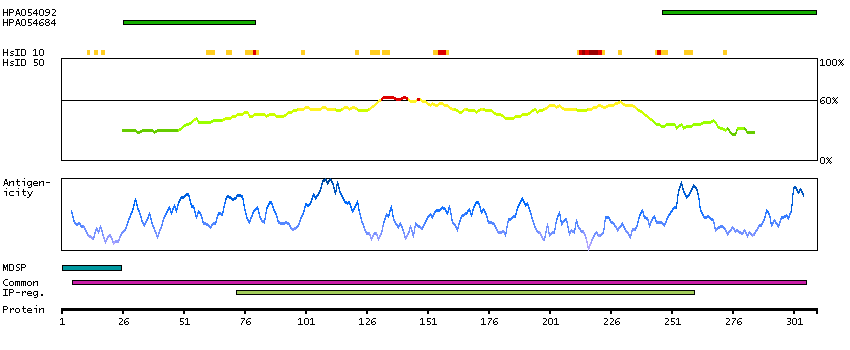
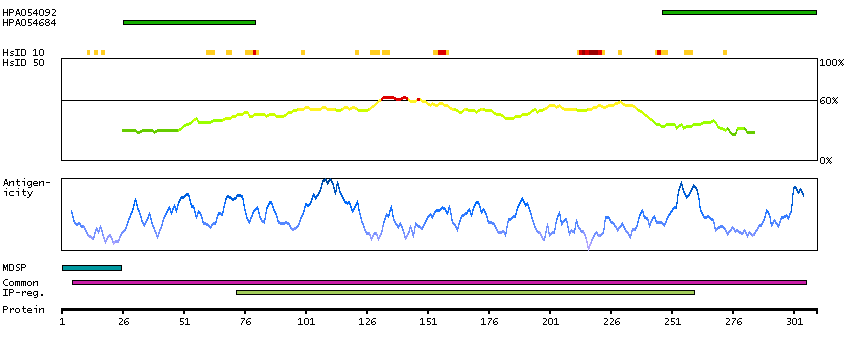
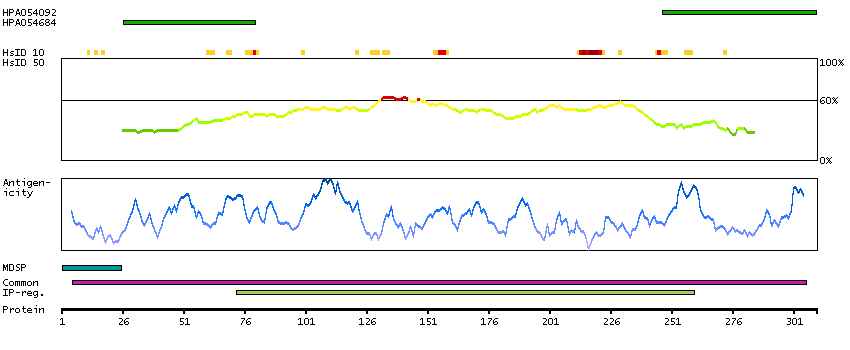
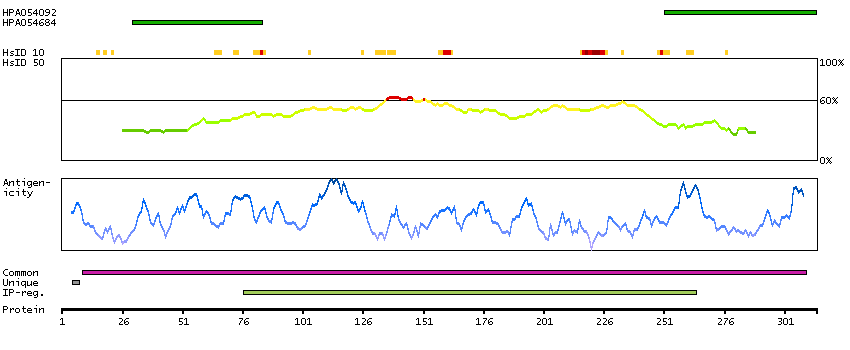
This gene encodes a member of a family of membrane-bound glycoproteins. The encoded protein may synthesize type 1 Lewis antigens, which are elevated in gastrointestinal and pancreatic cancers. Alternatively spliced transcript variants have been observed for this gene, but the full-length nature of some of these variants has not been determined. [provided by RefSeq, Jul 2013]