We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
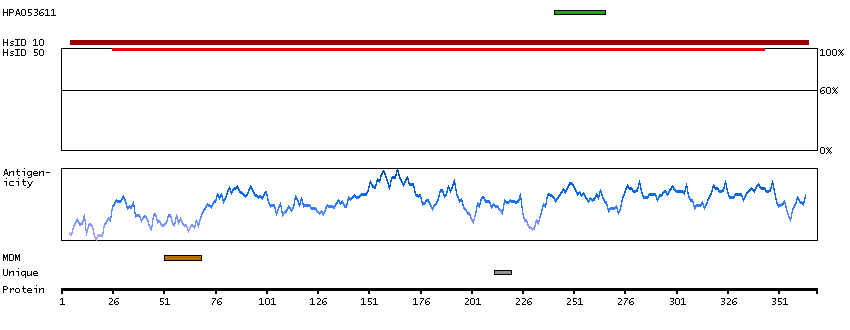
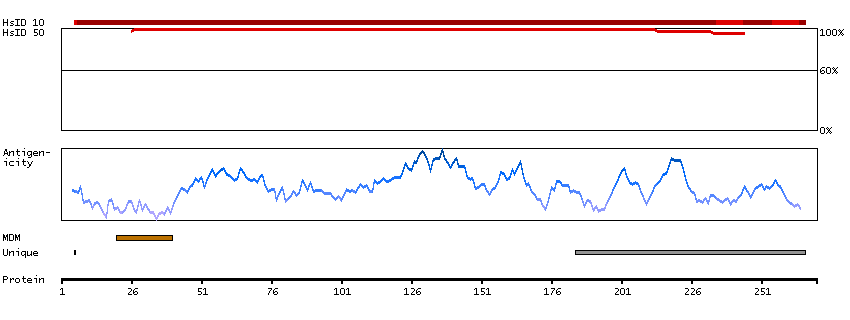
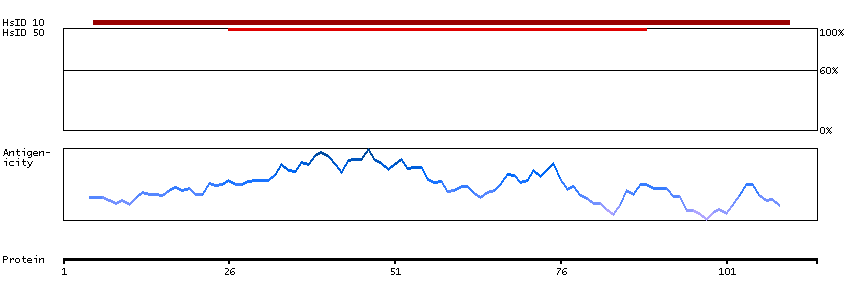
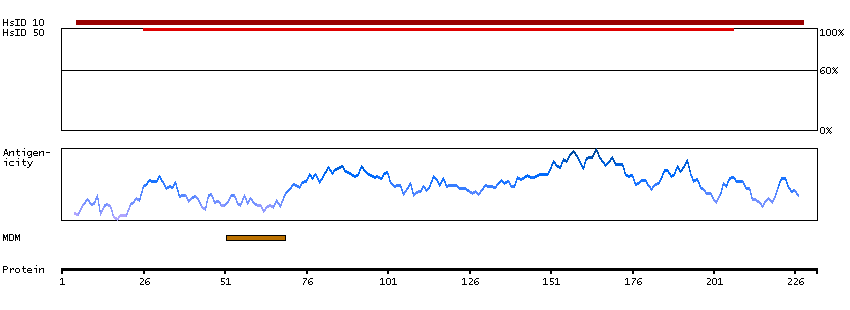
Q9NRE7 [Direct mapping] KIAA0220-like protein; Nuclear pore complex-interacting protein family member A5; Protein LOC102724993; Uncharacterized protein