We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
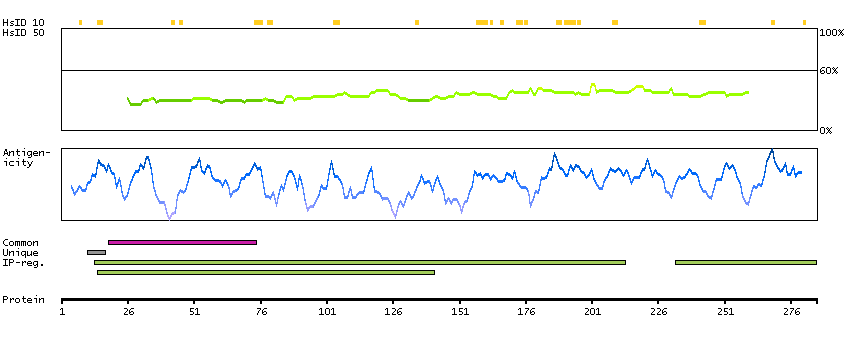
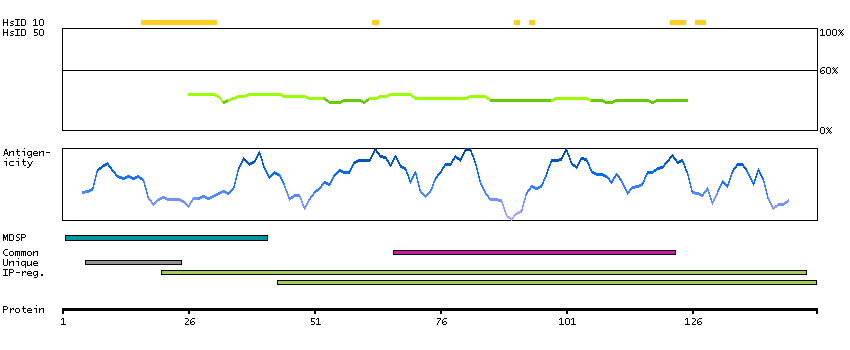
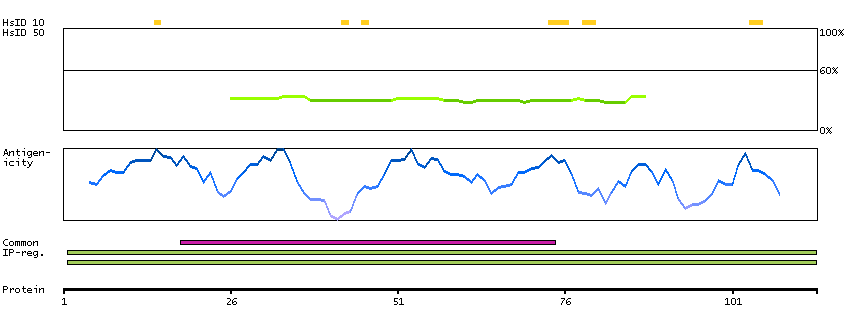
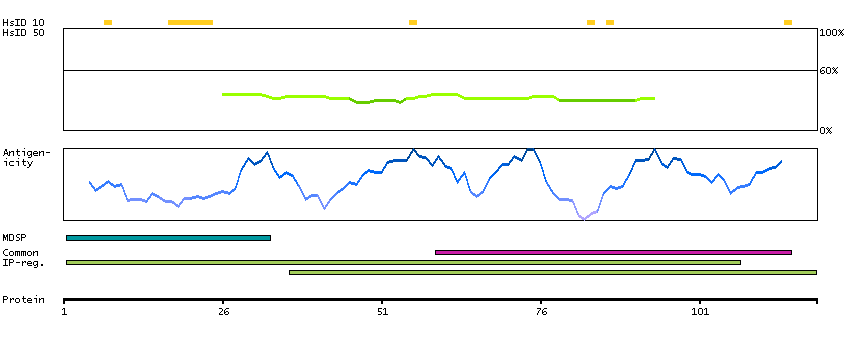
The protein encoded by this gene is a cytokine that controls the production, differentiation, and function of macrophages. The active form of the protein is found extracellularly as a disulfide-linked homodimer, and is thought to be produced by proteolytic cleavage of membrane-bound precursors. The encoded protein may be involved in development of the placenta. Alternate splicing results in multiple transcript variants. [provided by RefSeq, Sep 2011]