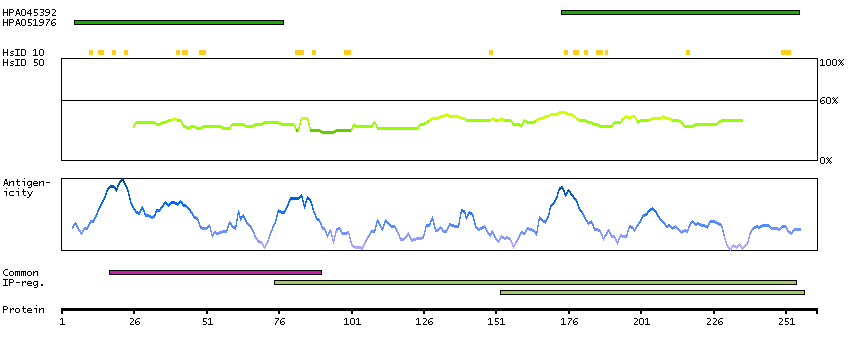
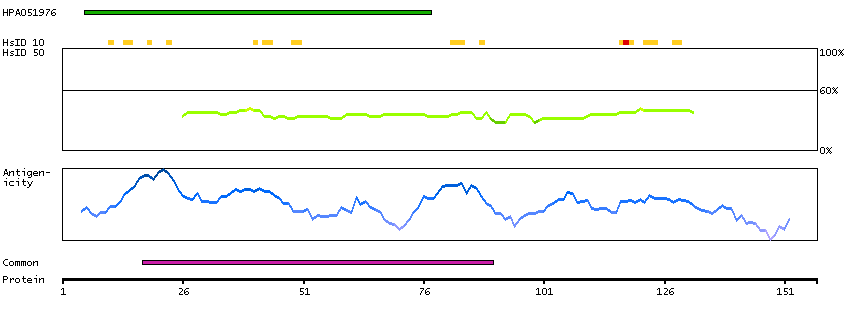
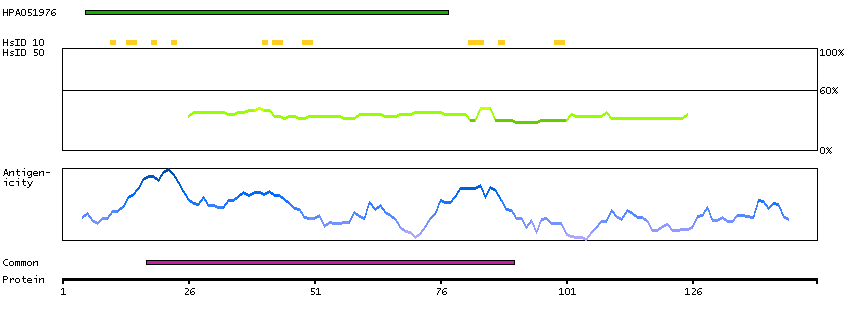
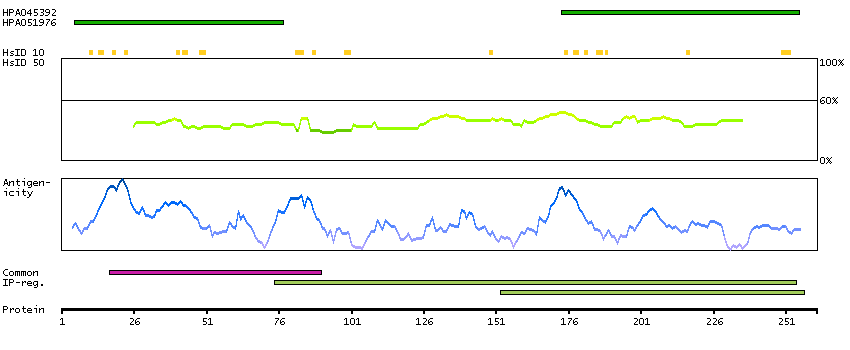
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
The Escherichia coli AlkB protein protects against the cytotoxicity of methylating agents by repair of the specific DNA lesions generated in single-stranded DNA. ALKBH2 and ALKBH3 (MIM 610603) are E. coli AlkB homologs that catalyze the removal of 1-methyladenine and 3-methylcytosine (Duncan et al., 2002 [PubMed 12486230]).[supplied by OMIM, Mar 2008]
Enzymes ENZYME proteins Oxidoreductases Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0006281 [DNA repair] GO:0006307 [DNA dealkylation involved in DNA repair] GO:0008198 [ferrous iron binding] GO:0015630 [microtubule cytoskeleton] GO:0016491 [oxidoreductase activity] GO:0016706 [oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, 2-oxoglutarate as one donor, and incorporation of one atom each of oxygen into both donors] GO:0035511 [oxidative DNA demethylation] GO:0043734 [DNA-N1-methyladenine dioxygenase activity] GO:0051747 [cytosine C-5 DNA demethylase activity] GO:0055114 [oxidation-reduction process] GO:0070989 [oxidative demethylation] GO:0080111 [DNA demethylation]