We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
This gene encodes an enzyme that catalyzes the hydrolysis of adenosine to inosine. Various mutations have been described for this gene and have been linked to human diseases. Deficiency in this enzyme causes a form of severe combined immunodeficiency disease (SCID), in which there is dysfunction of both B and T lymphocytes with impaired cellular immunity and decreased production of immunoglobulins, whereas elevated levels of this enzyme have been associated with congenital hemolytic anemia. [provided by RefSeq, Jul 2008]
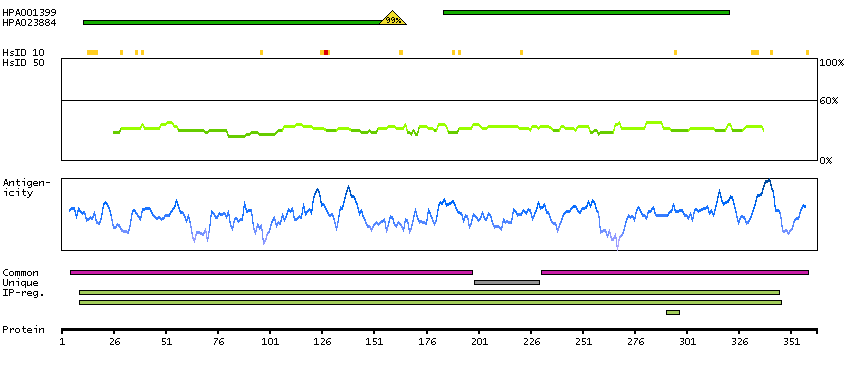
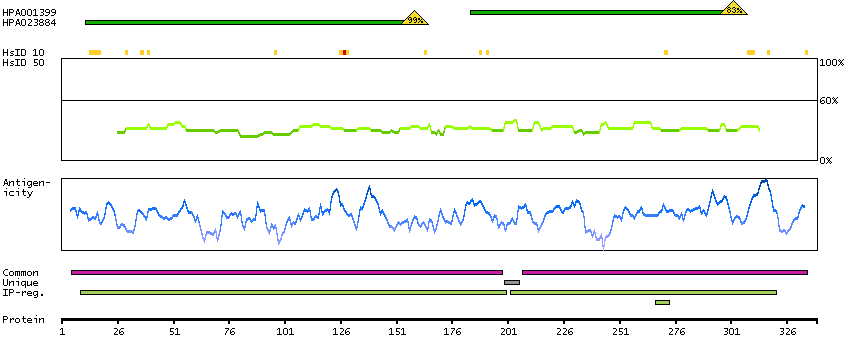
Enzymes ENZYME proteins Hydrolases Predicted intracellular proteins Plasma proteins Disease related genes FDA approved drug targets Small molecule drugs Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0001666 [response to hypoxia] GO:0001701 [in utero embryonic development] GO:0001821 [histamine secretion] GO:0001829 [trophectodermal cell differentiation] GO:0001883 [purine nucleoside binding] GO:0001889 [liver development] GO:0001890 [placenta development] GO:0002314 [germinal center B cell differentiation] GO:0002636 [positive regulation of germinal center formation] GO:0002686 [negative regulation of leukocyte migration] GO:0002906 [negative regulation of mature B cell apoptotic process] GO:0004000 [adenosine deaminase activity] GO:0005515 [protein binding] GO:0005615 [extracellular space] GO:0005737 [cytoplasm] GO:0005764 [lysosome] GO:0005829 [cytosol] GO:0005886 [plasma membrane] GO:0006144 [purine nucleobase metabolic process] GO:0006154 [adenosine catabolic process] GO:0006157 [deoxyadenosine catabolic process] GO:0007568 [aging] GO:0008270 [zinc ion binding] GO:0009168 [purine ribonucleoside monophosphate biosynthetic process] GO:0009897 [external side of plasma membrane] GO:0009986 [cell surface] GO:0010460 [positive regulation of heart rate] GO:0016020 [membrane] GO:0019239 [deaminase activity] GO:0030054 [cell junction] GO:0030324 [lung development] GO:0030890 [positive regulation of B cell proliferation] GO:0032261 [purine nucleotide salvage] GO:0032839 [dendrite cytoplasm] GO:0033089 [positive regulation of T cell differentiation in thymus] GO:0033197 [response to vitamin E] GO:0033632 [regulation of cell-cell adhesion mediated by integrin] GO:0042110 [T cell activation] GO:0042323 [negative regulation of circadian sleep/wake cycle, non-REM sleep] GO:0042493 [response to drug] GO:0042542 [response to hydrogen peroxide] GO:0043025 [neuronal cell body] GO:0043066 [negative regulation of apoptotic process] GO:0043101 [purine-containing compound salvage] GO:0043103 [hypoxanthine salvage] GO:0043278 [response to morphine] GO:0044281 [small molecule metabolic process] GO:0045187 [regulation of circadian sleep/wake cycle, sleep] GO:0045580 [regulation of T cell differentiation] GO:0045582 [positive regulation of T cell differentiation] GO:0045987 [positive regulation of smooth muscle contraction] GO:0046061 [dATP catabolic process] GO:0046101 [hypoxanthine biosynthetic process] GO:0046103 [inosine biosynthetic process] GO:0046111 [xanthine biosynthetic process] GO:0046638 [positive regulation of alpha-beta T cell differentiation] GO:0048286 [lung alveolus development] GO:0048541 [Peyer's patch development] GO:0048566 [embryonic digestive tract development] GO:0050728 [negative regulation of inflammatory response] GO:0050850 [positive regulation of calcium-mediated signaling] GO:0050862 [positive regulation of T cell receptor signaling pathway] GO:0050870 [positive regulation of T cell activation] GO:0055086 [nucleobase-containing small molecule metabolic process] GO:0060169 [negative regulation of adenosine receptor signaling pathway] GO:0060205 [cytoplasmic membrane-bounded vesicle lumen] GO:0060407 [negative regulation of penile erection] GO:0070244 [negative regulation of thymocyte apoptotic process] GO:0070256 [negative regulation of mucus secretion]