We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
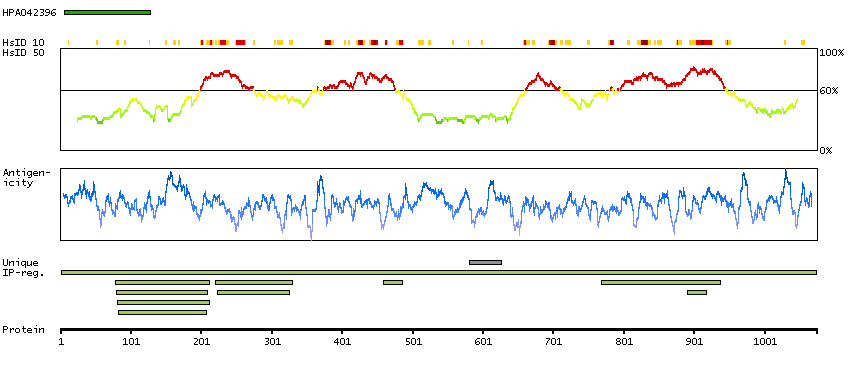
This gene encodes a phosphatidylcholine-specific phospholipase which catalyzes the hydrolysis of phosphatidylcholine in order to yield phosphatidic acid and choline. The enzyme may play a role in signal transduction and subcellular trafficking. Alternative splicing results in multiple transcript variants with both catalytic and regulatory properties. [provided by RefSeq, Sep 2011]
Enzymes ENZYME proteins Hydrolases Predicted intracellular proteins Plasma proteins RAS pathway related proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Hydrolases Predicted intracellular proteins Plasma proteins RAS pathway related proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)