We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
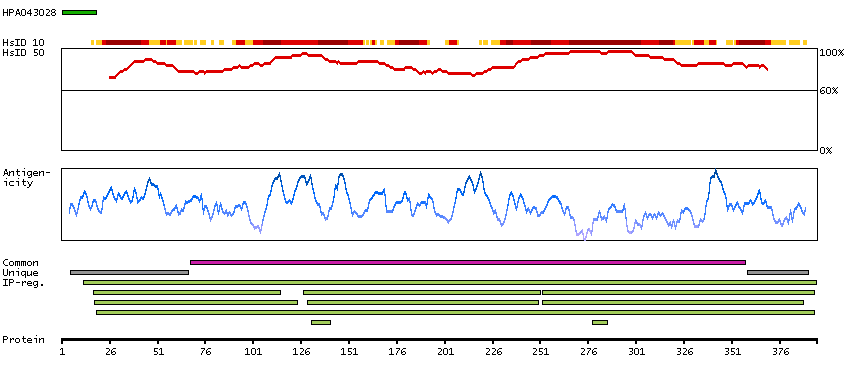
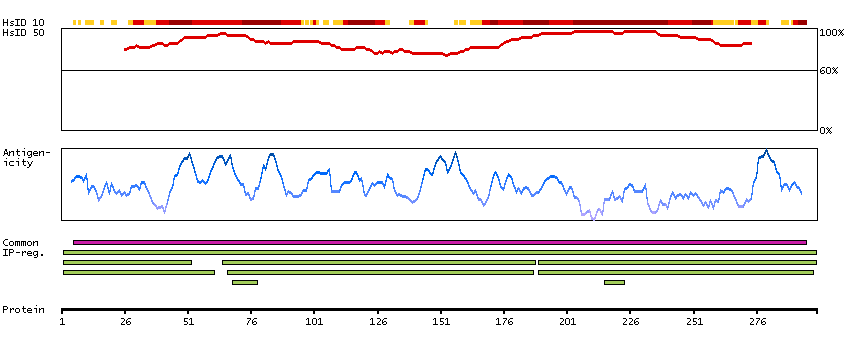
Methionine adenosyltransferase II, alpha (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene catalyzes the production of S-adenosylmethionine (AdoMet) from methionine and ATP. AdoMet is the key methyl donor in cellular processes. [provided by RefSeq, Jun 2011]
Enzymes ENZYME proteins Transferases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)