We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
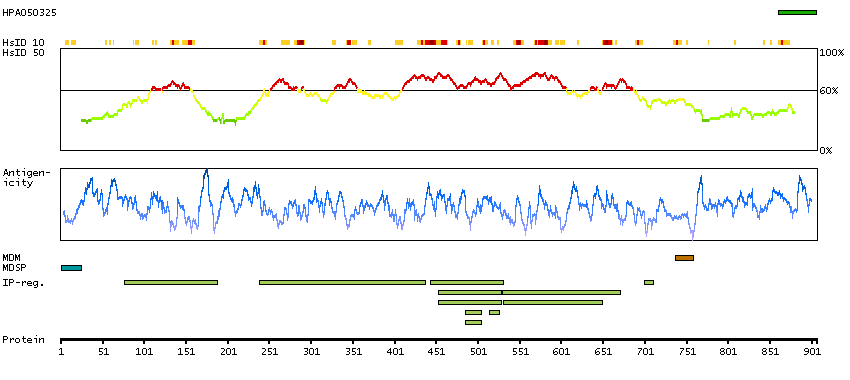
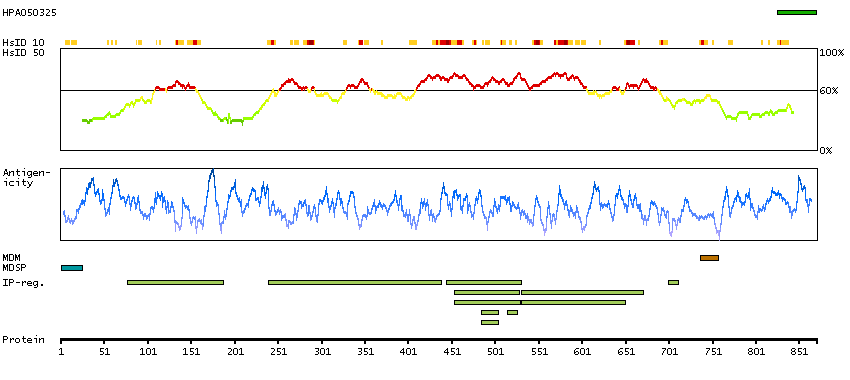
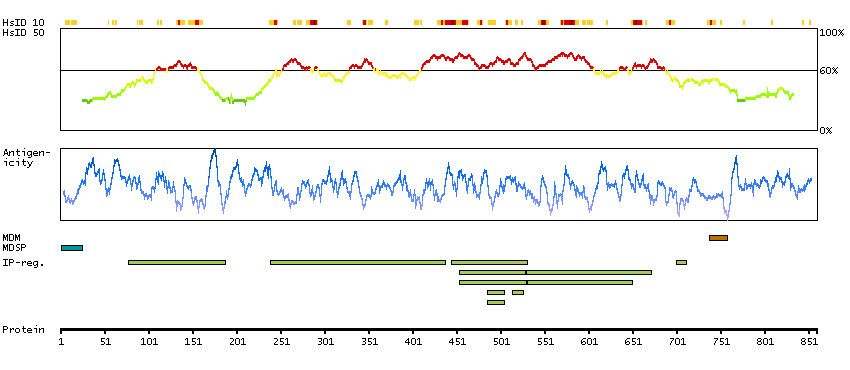
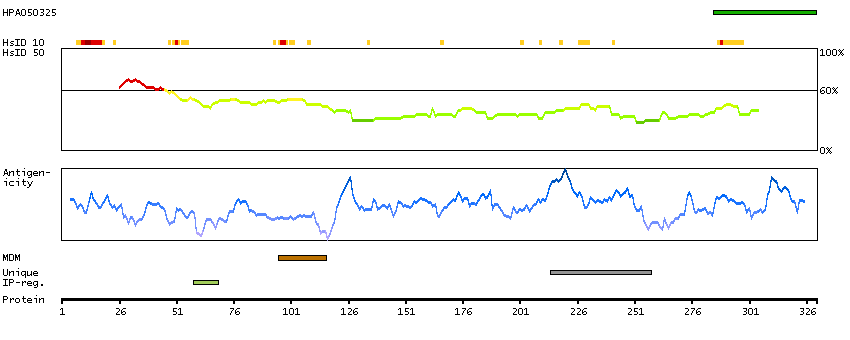
This gene encodes a member of the ADAM (a disintegrin and metalloprotease domain) family. Members of this family are membrane-anchored proteins structurally related to snake venom disintegrins, and have been implicated in a variety of biological processes involving cell-cell and cell-matrix interactions, including fertilization, muscle development, and neurogenesis. Unlike other members of the ADAM protein family, the protein encoded by this gene lacks metalloprotease activity since it has no zinc-binding motif. This gene is highly expressed in the brain and may function as an integrin ligand in the brain. In mice, it has been shown to be essential for correct myelination in the peripheral nervous system. Alternative splicing results in several transcript variants.[provided by RefSeq, Dec 2010]