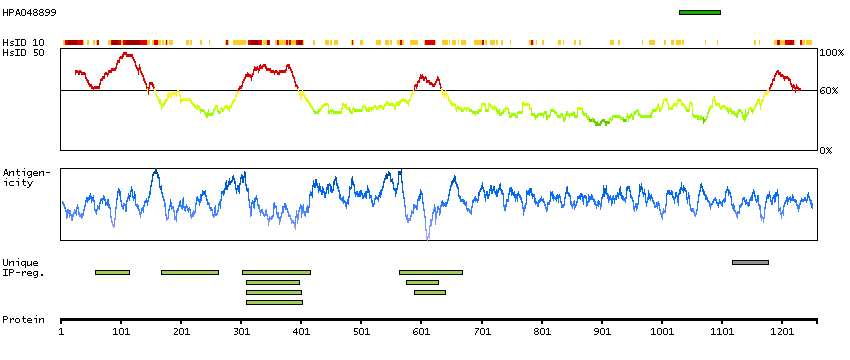
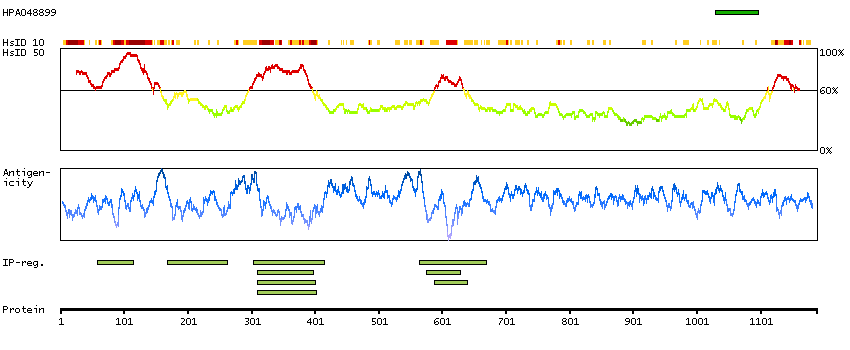
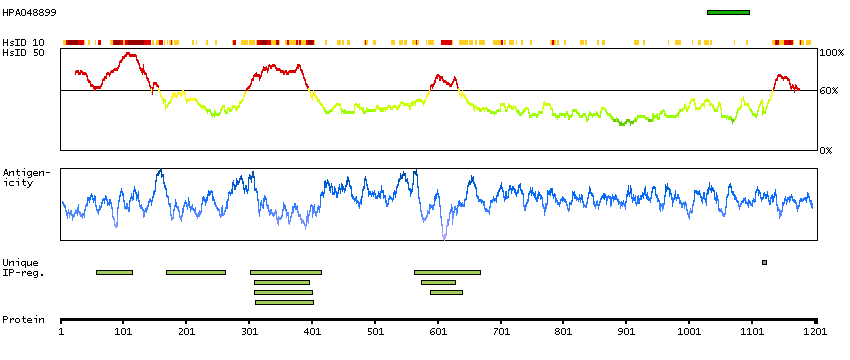
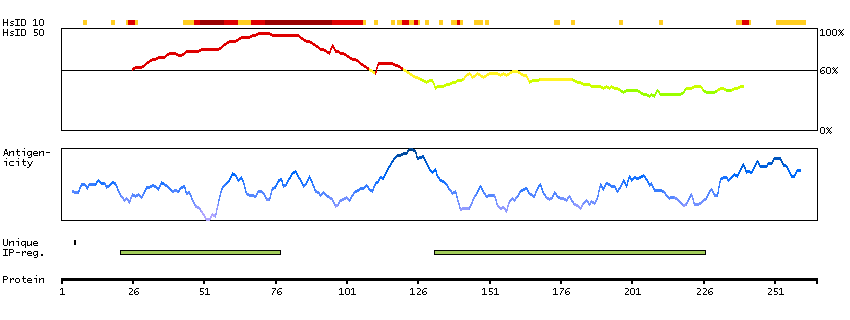
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
AT rich interactive domain 4A (RBP1-like) (HGNC Symbol)
Entrez gene summary
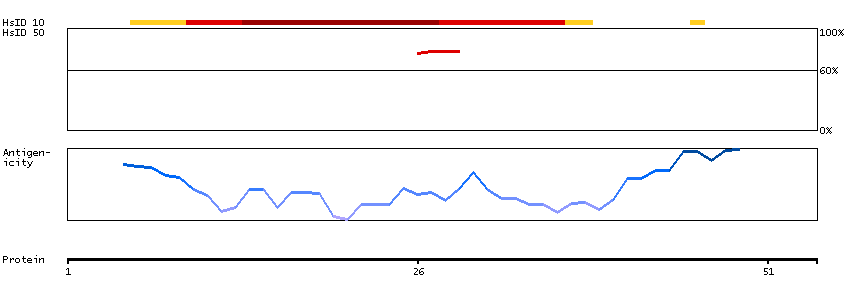
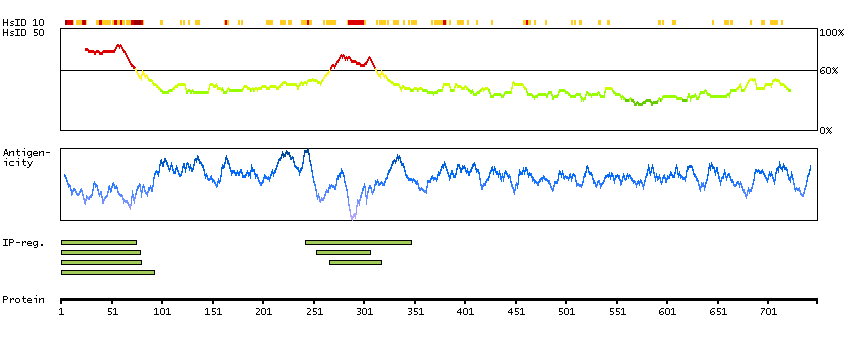
The protein encoded by this gene is a ubiquitously expressed nuclear protein. It binds directly, with several other proteins, to retinoblastoma protein (pRB) which regulates cell proliferation. pRB represses transcription by recruiting the encoded protein. This protein, in turn, serves as a bridging molecule to recruit HDACs and, in addition, provides a second HDAC-independent repression function. The encoded protein possesses transcriptional repression activity. Multiple alternatively spliced transcripts have been observed for this gene, although not all transcript variants have been fully described. [provided by RefSeq, Jul 2008]
Predicted intracellular proteins Transcription factors Helix-turn-helix domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0006325 [chromatin organization] GO:0006349 [regulation of gene expression by genetic imprinting] GO:0006357 [regulation of transcription from RNA polymerase II promoter] GO:0006366 [transcription from RNA polymerase II promoter] GO:0007283 [spermatogenesis] GO:0017053 [transcriptional repressor complex] GO:0034773 [histone H4-K20 trimethylation] GO:0036124 [histone H3-K9 trimethylation] GO:0044212 [transcription regulatory region DNA binding] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0048821 [erythrocyte development] GO:0080182 [histone H3-K4 trimethylation] GO:0097368 [establishment of Sertoli cell barrier]
Predicted intracellular proteins Transcription factors Helix-turn-helix domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0006325 [chromatin organization] GO:0006357 [regulation of transcription from RNA polymerase II promoter] GO:0006366 [transcription from RNA polymerase II promoter] GO:0017053 [transcriptional repressor complex] GO:0044212 [transcription regulatory region DNA binding] GO:0045892 [negative regulation of transcription, DNA-templated]
H7C485 [Direct mapping] AT-rich interactive domain-containing protein 4A
Show all
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003677 [DNA binding] GO:0005634 [nucleus] GO:0006357 [regulation of transcription from RNA polymerase II promoter] GO:0044212 [transcription regulatory region DNA binding]
Predicted intracellular proteins Transcription factors Helix-turn-helix domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0006325 [chromatin organization] GO:0006357 [regulation of transcription from RNA polymerase II promoter] GO:0006366 [transcription from RNA polymerase II promoter] GO:0017053 [transcriptional repressor complex] GO:0044212 [transcription regulatory region DNA binding] GO:0045892 [negative regulation of transcription, DNA-templated]