We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
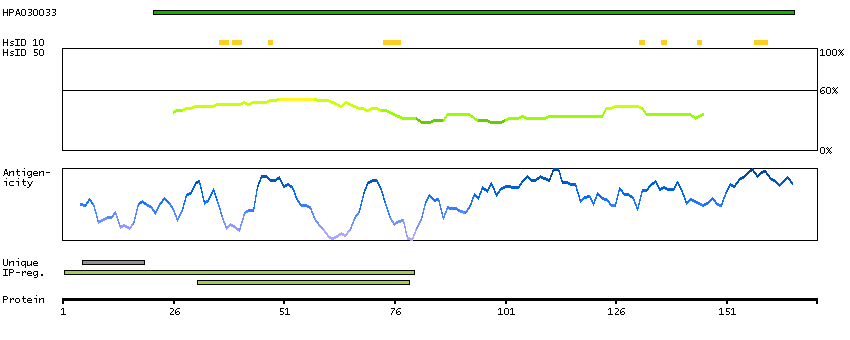
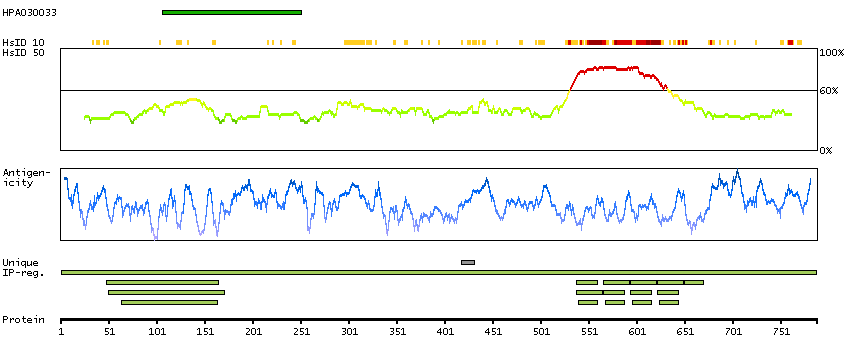
PR domain containing 1, with ZNF domain (HGNC Symbol)
Entrez gene summary
This gene encodes a protein that acts as a repressor of beta-interferon gene expression. The protein binds specifically to the PRDI (positive regulatory domain I element) of the beta-IFN gene promoter. Transcription of this gene increases upon virus induction. Two alternatively spliced transcript variants that encode different isoforms have been reported. [provided by RefSeq, Jul 2008]