We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Mitogen-activated protein kinase kinase 4 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the mitogen-activated protein kinase (MAPK) family. Members of this family act as an integration point for multiple biochemical signals and are involved in a wide variety of cellular processes such as proliferation, differentiation, transcription regulation, and development. They form a three-tiered signaling module composed of MAPKKKs, MAPKKs, and MAPKs. This protein is phosphorylated at serine and threonine residues by MAPKKKs and subsequently phosphorylates downstream MAPK targets at threonine and tyrosine residues. A similar protein in mouse has been reported to play a role in liver organogenesis. A pseudogene of this gene is located on the long arm of chromosome X. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jul 2013]
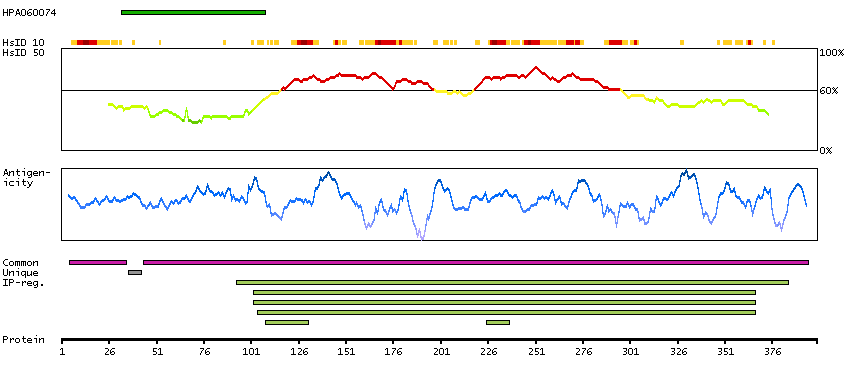
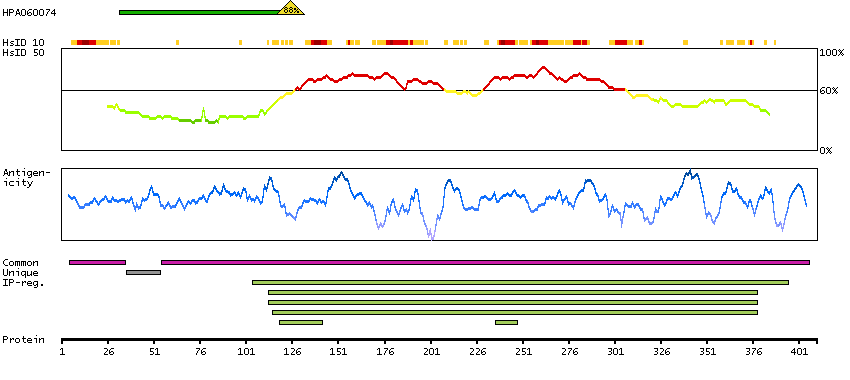
P45985 [Direct mapping] Dual specificity mitogen-activated protein kinase kinase 4
Show all
Enzymes ENZYME proteins Transferases Kinases STE Ser/Thr protein kinases SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Plasma proteins Cancer-related genes Candidate cancer biomarkers Mutated cancer genes Mutational cancer driver genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Nonsense Mutations COSMIC Missense Mutations COSMIC Large Deletions Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)