We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
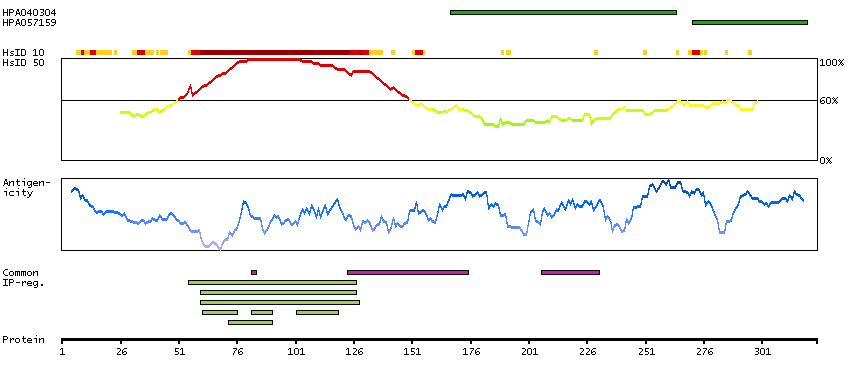
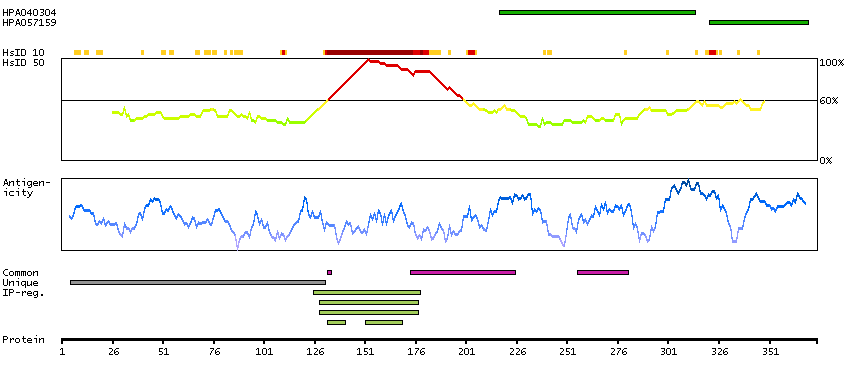
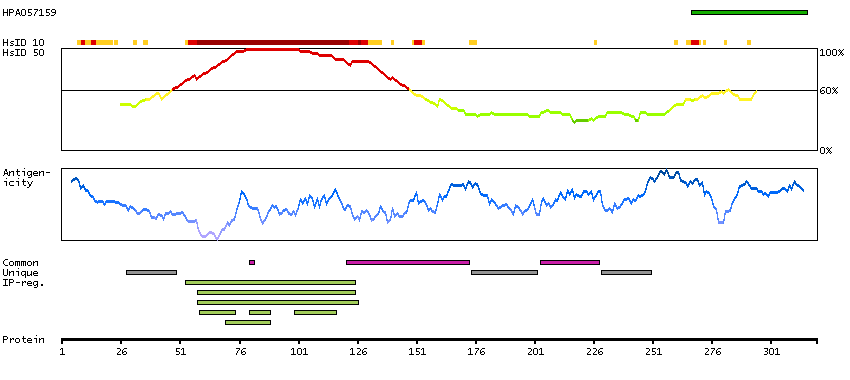
Antibody staining mainly consistent with RNA expression data. Secreted protein, tissue location of RNA and protein might differ and correlation is complex.
This gene encodes a highly conserved cold shock domain protein that has broad nucleic acid binding properties. The encoded protein functions as both a DNA and RNA binding protein and has been implicated in numerous cellular processes including regulation of transcription and translation, pre-mRNA splicing, DNA reparation and mRNA packaging. This protein is also a component of messenger ribonucleoprotein (mRNP) complexes and may have a role in microRNA processing. This protein can be secreted through non-classical pathways and functions as an extracellular mitogen. Aberrant expression of the gene is associated with cancer proliferation in numerous tissues. This gene may be a prognostic marker for poor outcome and drug resistance in certain cancers. Alternate splicing results in multiple transcript variants. Pseudogenes of this gene are found on multiple chromosomes. [provided by RefSeq, Sep 2015]
P67809 [Direct mapping] Nuclease-sensitive element-binding protein 1
Show all
Predicted intracellular proteins Plasma proteins Transcription factors beta-Barrel DNA-binding domains Cancer-related genes Candidate cancer biomarkers Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000398 [mRNA splicing, via spliceosome] GO:0000978 [RNA polymerase II core promoter proximal region sequence-specific DNA binding] GO:0001077 [transcriptional activator activity, RNA polymerase II core promoter proximal region sequence-specific binding] GO:0001701 [in utero embryonic development] GO:0002039 [p53 binding] GO:0003676 [nucleic acid binding] GO:0003677 [DNA binding] GO:0003682 [chromatin binding] GO:0003690 [double-stranded DNA binding] GO:0003697 [single-stranded DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0003723 [RNA binding] GO:0003729 [mRNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005689 [U12-type spliceosomal complex] GO:0005737 [cytoplasm] GO:0006355 [regulation of transcription, DNA-templated] GO:0006366 [transcription from RNA polymerase II promoter] GO:0008134 [transcription factor binding] GO:0008284 [positive regulation of cell proliferation] GO:0008380 [RNA splicing] GO:0010467 [gene expression] GO:0010494 [cytoplasmic stress granule] GO:0030425 [dendrite] GO:0030529 [ribonucleoprotein complex] GO:0031965 [nuclear membrane] GO:0043066 [negative regulation of apoptotic process] GO:0043231 [intracellular membrane-bounded organelle] GO:0043565 [sequence-specific DNA binding] GO:0044822 [poly(A) RNA binding] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046627 [negative regulation of insulin receptor signaling pathway] GO:0048471 [perinuclear region of cytoplasm] GO:0051020 [GTPase binding] GO:0051154 [negative regulation of striated muscle cell differentiation] GO:0051781 [positive regulation of cell division] GO:0070062 [extracellular exosome] GO:0070934 [CRD-mediated mRNA stabilization] GO:0070937 [CRD-mediated mRNA stability complex] GO:0071204 [histone pre-mRNA 3'end processing complex] GO:1990124 [messenger ribonucleoprotein complex]