We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
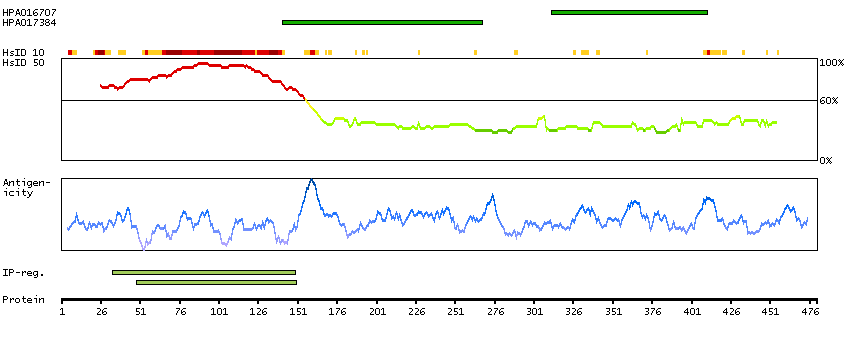
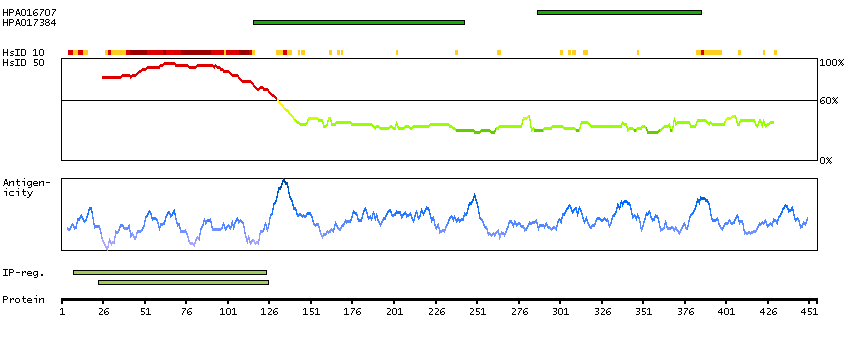
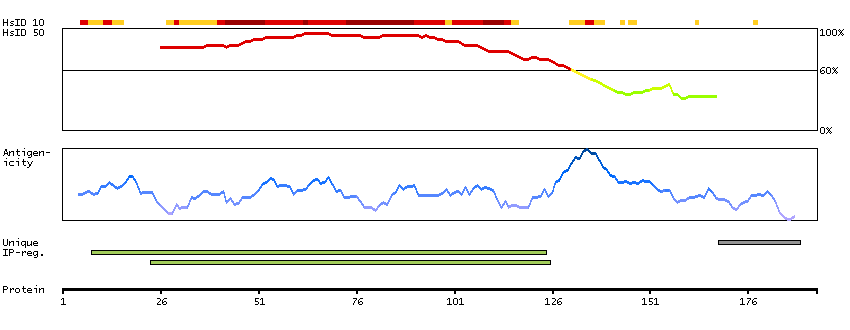
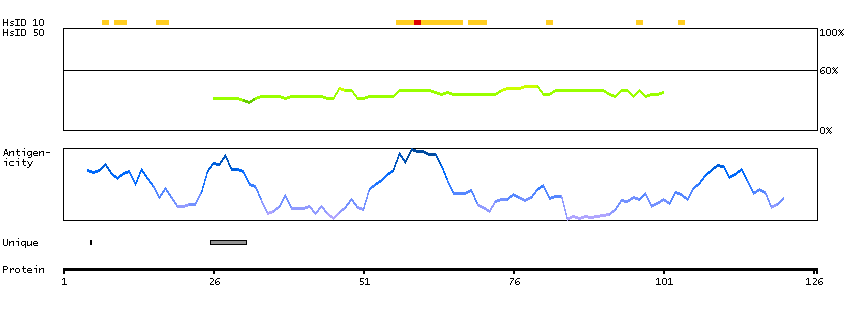
This gene encodes a subnuclear organelle and major component of the PML (promyelocytic leukemia)-SP100 nuclear bodies. PML and SP100 are covalently modified by the SUMO-1 modifier, which is considered crucial to nuclear body interactions. The encoded protein binds heterochromatin proteins and is thought to play a role in tumorigenesis, immunity, and gene regulation. Alternatively spliced variants have been identified for this gene; one of which encodes a high-mobility group protein. [provided by RefSeq, Aug 2011]
Predicted intracellular proteins Transcription factors alpha-Helices exposed by beta-structures Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000723 [telomere maintenance] GO:0000784 [nuclear chromosome, telomeric region] GO:0003677 [DNA binding] GO:0003682 [chromatin binding] GO:0003713 [transcription coactivator activity] GO:0003714 [transcription corepressor activity] GO:0005125 [cytokine activity] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0006338 [chromatin remodeling] GO:0006351 [transcription, DNA-templated] GO:0006954 [inflammatory response] GO:0006978 [DNA damage response, signal transduction by p53 class mediator resulting in transcription of p21 class mediator] GO:0008134 [transcription factor binding] GO:0010596 [negative regulation of endothelial cell migration] GO:0016032 [viral process] GO:0016605 [PML body] GO:0016925 [protein sumoylation] GO:0019221 [cytokine-mediated signaling pathway] GO:0019900 [kinase binding] GO:0019904 [protein domain specific binding] GO:0030870 [Mre11 complex] GO:0032526 [response to retinoic acid] GO:0032897 [negative regulation of viral transcription] GO:0034097 [response to cytokine] GO:0034340 [response to type I interferon] GO:0034341 [response to interferon-gamma] GO:0034399 [nuclear periphery] GO:0042802 [identical protein binding] GO:0042803 [protein homodimerization activity] GO:0043392 [negative regulation of DNA binding] GO:0043433 [negative regulation of sequence-specific DNA binding transcription factor activity] GO:0043687 [post-translational protein modification] GO:0044267 [cellular protein metabolic process] GO:0045765 [regulation of angiogenesis] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0046826 [negative regulation of protein export from nucleus] GO:0048384 [retinoic acid receptor signaling pathway] GO:0051091 [positive regulation of sequence-specific DNA binding transcription factor activity] GO:0051271 [negative regulation of cellular component movement] GO:0060333 [interferon-gamma-mediated signaling pathway] GO:0060337 [type I interferon signaling pathway] GO:0070087 [chromo shadow domain binding] GO:1902041 [regulation of extrinsic apoptotic signaling pathway via death domain receptors] GO:1902044 [regulation of Fas signaling pathway] GO:1903507 [negative regulation of nucleic acid-templated transcription]
Predicted intracellular proteins Transcription factors alpha-Helices exposed by beta-structures Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000723 [telomere maintenance] GO:0000784 [nuclear chromosome, telomeric region] GO:0003677 [DNA binding] GO:0003682 [chromatin binding] GO:0003713 [transcription coactivator activity] GO:0003714 [transcription corepressor activity] GO:0005125 [cytokine activity] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0006338 [chromatin remodeling] GO:0006351 [transcription, DNA-templated] GO:0006954 [inflammatory response] GO:0006978 [DNA damage response, signal transduction by p53 class mediator resulting in transcription of p21 class mediator] GO:0008134 [transcription factor binding] GO:0008270 [zinc ion binding] GO:0010596 [negative regulation of endothelial cell migration] GO:0016032 [viral process] GO:0016605 [PML body] GO:0016925 [protein sumoylation] GO:0019221 [cytokine-mediated signaling pathway] GO:0019900 [kinase binding] GO:0019904 [protein domain specific binding] GO:0030870 [Mre11 complex] GO:0032526 [response to retinoic acid] GO:0032897 [negative regulation of viral transcription] GO:0034097 [response to cytokine] GO:0034340 [response to type I interferon] GO:0034341 [response to interferon-gamma] GO:0034399 [nuclear periphery] GO:0042802 [identical protein binding] GO:0042803 [protein homodimerization activity] GO:0043392 [negative regulation of DNA binding] GO:0043433 [negative regulation of sequence-specific DNA binding transcription factor activity] GO:0043687 [post-translational protein modification] GO:0044267 [cellular protein metabolic process] GO:0045765 [regulation of angiogenesis] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0046826 [negative regulation of protein export from nucleus] GO:0048384 [retinoic acid receptor signaling pathway] GO:0051091 [positive regulation of sequence-specific DNA binding transcription factor activity] GO:0051271 [negative regulation of cellular component movement] GO:0060333 [interferon-gamma-mediated signaling pathway] GO:0060337 [type I interferon signaling pathway] GO:0070087 [chromo shadow domain binding] GO:1902041 [regulation of extrinsic apoptotic signaling pathway via death domain receptors] GO:1902044 [regulation of Fas signaling pathway] GO:1903507 [negative regulation of nucleic acid-templated transcription]
Predicted intracellular proteins Transcription factors alpha-Helices exposed by beta-structures Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000723 [telomere maintenance] GO:0000784 [nuclear chromosome, telomeric region] GO:0003677 [DNA binding] GO:0003682 [chromatin binding] GO:0003713 [transcription coactivator activity] GO:0003714 [transcription corepressor activity] GO:0005125 [cytokine activity] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0006338 [chromatin remodeling] GO:0006351 [transcription, DNA-templated] GO:0006954 [inflammatory response] GO:0006978 [DNA damage response, signal transduction by p53 class mediator resulting in transcription of p21 class mediator] GO:0008134 [transcription factor binding] GO:0010596 [negative regulation of endothelial cell migration] GO:0016032 [viral process] GO:0016605 [PML body] GO:0016925 [protein sumoylation] GO:0019221 [cytokine-mediated signaling pathway] GO:0019900 [kinase binding] GO:0019904 [protein domain specific binding] GO:0030870 [Mre11 complex] GO:0032526 [response to retinoic acid] GO:0032897 [negative regulation of viral transcription] GO:0034097 [response to cytokine] GO:0034340 [response to type I interferon] GO:0034341 [response to interferon-gamma] GO:0034399 [nuclear periphery] GO:0042802 [identical protein binding] GO:0042803 [protein homodimerization activity] GO:0043392 [negative regulation of DNA binding] GO:0043433 [negative regulation of sequence-specific DNA binding transcription factor activity] GO:0043687 [post-translational protein modification] GO:0044267 [cellular protein metabolic process] GO:0045765 [regulation of angiogenesis] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0046826 [negative regulation of protein export from nucleus] GO:0048384 [retinoic acid receptor signaling pathway] GO:0051091 [positive regulation of sequence-specific DNA binding transcription factor activity] GO:0051271 [negative regulation of cellular component movement] GO:0060333 [interferon-gamma-mediated signaling pathway] GO:0060337 [type I interferon signaling pathway] GO:0070087 [chromo shadow domain binding] GO:1902041 [regulation of extrinsic apoptotic signaling pathway via death domain receptors] GO:1902044 [regulation of Fas signaling pathway] GO:1903507 [negative regulation of nucleic acid-templated transcription]
Predicted intracellular proteins Transcription factors alpha-Helices exposed by beta-structures Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000723 [telomere maintenance] GO:0000784 [nuclear chromosome, telomeric region] GO:0003677 [DNA binding] GO:0003682 [chromatin binding] GO:0003713 [transcription coactivator activity] GO:0003714 [transcription corepressor activity] GO:0005125 [cytokine activity] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0006338 [chromatin remodeling] GO:0006351 [transcription, DNA-templated] GO:0006954 [inflammatory response] GO:0006978 [DNA damage response, signal transduction by p53 class mediator resulting in transcription of p21 class mediator] GO:0008134 [transcription factor binding] GO:0010596 [negative regulation of endothelial cell migration] GO:0016032 [viral process] GO:0016605 [PML body] GO:0016925 [protein sumoylation] GO:0019221 [cytokine-mediated signaling pathway] GO:0019900 [kinase binding] GO:0019904 [protein domain specific binding] GO:0030870 [Mre11 complex] GO:0032526 [response to retinoic acid] GO:0032897 [negative regulation of viral transcription] GO:0034097 [response to cytokine] GO:0034340 [response to type I interferon] GO:0034341 [response to interferon-gamma] GO:0034399 [nuclear periphery] GO:0042802 [identical protein binding] GO:0042803 [protein homodimerization activity] GO:0043392 [negative regulation of DNA binding] GO:0043433 [negative regulation of sequence-specific DNA binding transcription factor activity] GO:0043687 [post-translational protein modification] GO:0044267 [cellular protein metabolic process] GO:0045765 [regulation of angiogenesis] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0046826 [negative regulation of protein export from nucleus] GO:0048384 [retinoic acid receptor signaling pathway] GO:0051091 [positive regulation of sequence-specific DNA binding transcription factor activity] GO:0051271 [negative regulation of cellular component movement] GO:0060333 [interferon-gamma-mediated signaling pathway] GO:0060337 [type I interferon signaling pathway] GO:0070087 [chromo shadow domain binding] GO:1902041 [regulation of extrinsic apoptotic signaling pathway via death domain receptors] GO:1902044 [regulation of Fas signaling pathway] GO:1903507 [negative regulation of nucleic acid-templated transcription]
Predicted intracellular proteins Transcription factors alpha-Helices exposed by beta-structures Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000723 [telomere maintenance] GO:0000784 [nuclear chromosome, telomeric region] GO:0003677 [DNA binding] GO:0003682 [chromatin binding] GO:0003713 [transcription coactivator activity] GO:0003714 [transcription corepressor activity] GO:0005125 [cytokine activity] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0006338 [chromatin remodeling] GO:0006351 [transcription, DNA-templated] GO:0006954 [inflammatory response] GO:0006978 [DNA damage response, signal transduction by p53 class mediator resulting in transcription of p21 class mediator] GO:0008134 [transcription factor binding] GO:0010596 [negative regulation of endothelial cell migration] GO:0016032 [viral process] GO:0016605 [PML body] GO:0016925 [protein sumoylation] GO:0019221 [cytokine-mediated signaling pathway] GO:0019900 [kinase binding] GO:0019904 [protein domain specific binding] GO:0030870 [Mre11 complex] GO:0032526 [response to retinoic acid] GO:0032897 [negative regulation of viral transcription] GO:0034097 [response to cytokine] GO:0034340 [response to type I interferon] GO:0034341 [response to interferon-gamma] GO:0034399 [nuclear periphery] GO:0042802 [identical protein binding] GO:0042803 [protein homodimerization activity] GO:0043392 [negative regulation of DNA binding] GO:0043433 [negative regulation of sequence-specific DNA binding transcription factor activity] GO:0043687 [post-translational protein modification] GO:0044267 [cellular protein metabolic process] GO:0045765 [regulation of angiogenesis] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0046826 [negative regulation of protein export from nucleus] GO:0048384 [retinoic acid receptor signaling pathway] GO:0051091 [positive regulation of sequence-specific DNA binding transcription factor activity] GO:0051271 [negative regulation of cellular component movement] GO:0060333 [interferon-gamma-mediated signaling pathway] GO:0060337 [type I interferon signaling pathway] GO:0070087 [chromo shadow domain binding] GO:1902041 [regulation of extrinsic apoptotic signaling pathway via death domain receptors] GO:1902044 [regulation of Fas signaling pathway] GO:1903507 [negative regulation of nucleic acid-templated transcription]
Predicted intracellular proteins Transcription factors alpha-Helices exposed by beta-structures Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000723 [telomere maintenance] GO:0000784 [nuclear chromosome, telomeric region] GO:0003677 [DNA binding] GO:0003682 [chromatin binding] GO:0003713 [transcription coactivator activity] GO:0003714 [transcription corepressor activity] GO:0005125 [cytokine activity] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0006338 [chromatin remodeling] GO:0006351 [transcription, DNA-templated] GO:0006954 [inflammatory response] GO:0006978 [DNA damage response, signal transduction by p53 class mediator resulting in transcription of p21 class mediator] GO:0008134 [transcription factor binding] GO:0010596 [negative regulation of endothelial cell migration] GO:0016032 [viral process] GO:0016605 [PML body] GO:0016925 [protein sumoylation] GO:0019221 [cytokine-mediated signaling pathway] GO:0019900 [kinase binding] GO:0019904 [protein domain specific binding] GO:0030870 [Mre11 complex] GO:0032526 [response to retinoic acid] GO:0032897 [negative regulation of viral transcription] GO:0034097 [response to cytokine] GO:0034340 [response to type I interferon] GO:0034341 [response to interferon-gamma] GO:0034399 [nuclear periphery] GO:0042802 [identical protein binding] GO:0042803 [protein homodimerization activity] GO:0043392 [negative regulation of DNA binding] GO:0043433 [negative regulation of sequence-specific DNA binding transcription factor activity] GO:0043687 [post-translational protein modification] GO:0044267 [cellular protein metabolic process] GO:0045765 [regulation of angiogenesis] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0046826 [negative regulation of protein export from nucleus] GO:0048384 [retinoic acid receptor signaling pathway] GO:0051091 [positive regulation of sequence-specific DNA binding transcription factor activity] GO:0051271 [negative regulation of cellular component movement] GO:0060333 [interferon-gamma-mediated signaling pathway] GO:0060337 [type I interferon signaling pathway] GO:0070087 [chromo shadow domain binding] GO:1902041 [regulation of extrinsic apoptotic signaling pathway via death domain receptors] GO:1902044 [regulation of Fas signaling pathway] GO:1903507 [negative regulation of nucleic acid-templated transcription]