We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
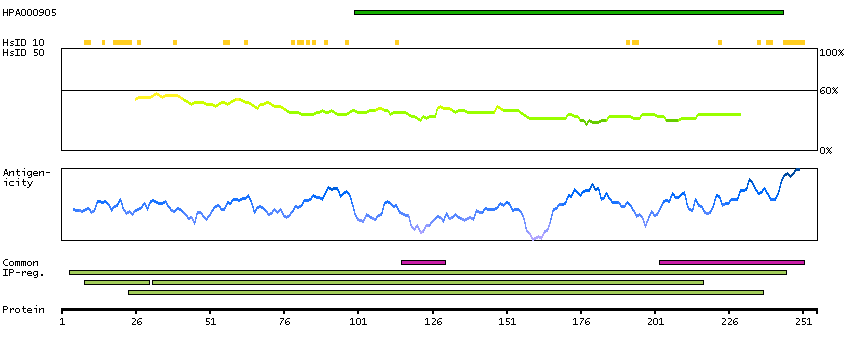
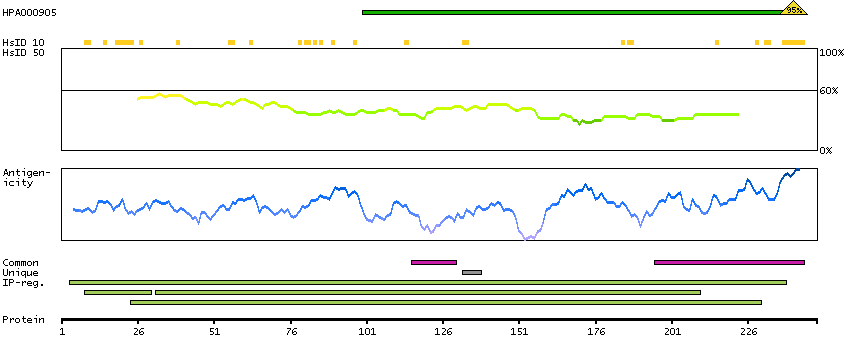
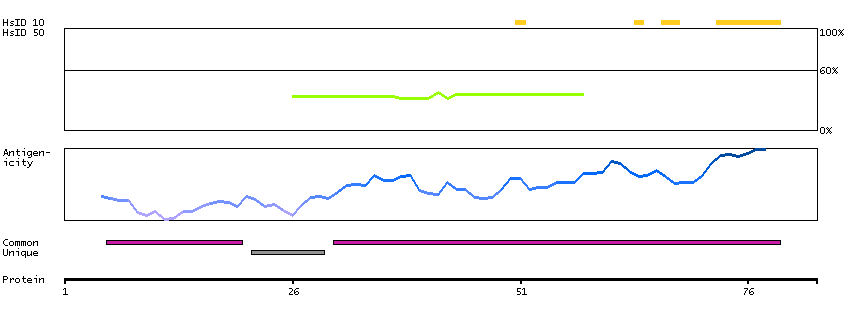
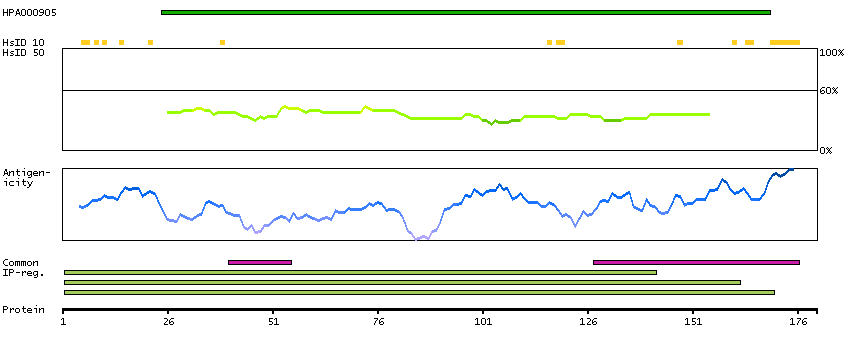
The proteasome is a multicatalytic proteinase complex with a highly ordered ring-shaped 20S core structure. The core structure is composed of 4 rings of 28 non-identical subunits; 2 rings are composed of 7 alpha subunits and 2 rings are composed of 7 beta subunits. Proteasomes are distributed throughout eukaryotic cells at a high concentration and cleave peptides in an ATP/ubiquitin-dependent process in a non-lysosomal pathway. An essential function of a modified proteasome, the immunoproteasome, is the processing of class I MHC peptides. This gene encodes a member of the peptidase T1A family, that is a 20S core alpha subunit. Two alternative transcripts encoding different isoforms have been identified. [provided by RefSeq, Jul 2008]
Enzymes ENZYME proteins Hydrolases Peptidases Threonine-type peptidases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000082 [G1/S transition of mitotic cell cycle] GO:0000165 [MAPK cascade] GO:0000186 [activation of MAPKK activity] GO:0000209 [protein polyubiquitination] GO:0000278 [mitotic cell cycle] GO:0000502 [proteasome complex] GO:0002223 [stimulatory C-type lectin receptor signaling pathway] GO:0002474 [antigen processing and presentation of peptide antigen via MHC class I] GO:0002479 [antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-dependent] GO:0004175 [endopeptidase activity] GO:0004298 [threonine-type endopeptidase activity] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005737 [cytoplasm] GO:0005829 [cytosol] GO:0005839 [proteasome core complex] GO:0006511 [ubiquitin-dependent protein catabolic process] GO:0006521 [regulation of cellular amino acid metabolic process] GO:0006595 [polyamine metabolic process] GO:0006915 [apoptotic process] GO:0006977 [DNA damage response, signal transduction by p53 class mediator resulting in cell cycle arrest] GO:0007173 [epidermal growth factor receptor signaling pathway] GO:0007264 [small GTPase mediated signal transduction] GO:0007265 [Ras protein signal transduction] GO:0007411 [axon guidance] GO:0008286 [insulin receptor signaling pathway] GO:0008543 [fibroblast growth factor receptor signaling pathway] GO:0010467 [gene expression] GO:0010499 [proteasomal ubiquitin-independent protein catabolic process] GO:0012501 [programmed cell death] GO:0016032 [viral process] GO:0019773 [proteasome core complex, alpha-subunit complex] GO:0031145 [anaphase-promoting complex-dependent proteasomal ubiquitin-dependent protein catabolic process] GO:0033209 [tumor necrosis factor-mediated signaling pathway] GO:0034641 [cellular nitrogen compound metabolic process] GO:0038061 [NIK/NF-kappaB signaling] GO:0038095 [Fc-epsilon receptor signaling pathway] GO:0042590 [antigen processing and presentation of exogenous peptide antigen via MHC class I] GO:0042981 [regulation of apoptotic process] GO:0043066 [negative regulation of apoptotic process] GO:0043161 [proteasome-mediated ubiquitin-dependent protein catabolic process] GO:0043488 [regulation of mRNA stability] GO:0044281 [small molecule metabolic process] GO:0045087 [innate immune response] GO:0048010 [vascular endothelial growth factor receptor signaling pathway] GO:0048011 [neurotrophin TRK receptor signaling pathway] GO:0050852 [T cell receptor signaling pathway] GO:0051436 [negative regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle] GO:0051437 [positive regulation of ubiquitin-protein ligase activity involved in regulation of mitotic cell cycle transition] GO:0051439 [regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle] GO:0051603 [proteolysis involved in cellular protein catabolic process] GO:0070062 [extracellular exosome] GO:0090090 [negative regulation of canonical Wnt signaling pathway] GO:0090263 [positive regulation of canonical Wnt signaling pathway]
Enzymes ENZYME proteins Hydrolases Peptidases Threonine-type peptidases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000082 [G1/S transition of mitotic cell cycle] GO:0000165 [MAPK cascade] GO:0000186 [activation of MAPKK activity] GO:0000209 [protein polyubiquitination] GO:0000278 [mitotic cell cycle] GO:0000502 [proteasome complex] GO:0002223 [stimulatory C-type lectin receptor signaling pathway] GO:0002474 [antigen processing and presentation of peptide antigen via MHC class I] GO:0002479 [antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-dependent] GO:0004175 [endopeptidase activity] GO:0004298 [threonine-type endopeptidase activity] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005737 [cytoplasm] GO:0005829 [cytosol] GO:0005839 [proteasome core complex] GO:0006511 [ubiquitin-dependent protein catabolic process] GO:0006521 [regulation of cellular amino acid metabolic process] GO:0006595 [polyamine metabolic process] GO:0006915 [apoptotic process] GO:0006977 [DNA damage response, signal transduction by p53 class mediator resulting in cell cycle arrest] GO:0007173 [epidermal growth factor receptor signaling pathway] GO:0007264 [small GTPase mediated signal transduction] GO:0007265 [Ras protein signal transduction] GO:0007411 [axon guidance] GO:0008286 [insulin receptor signaling pathway] GO:0008543 [fibroblast growth factor receptor signaling pathway] GO:0010467 [gene expression] GO:0010499 [proteasomal ubiquitin-independent protein catabolic process] GO:0012501 [programmed cell death] GO:0016032 [viral process] GO:0019773 [proteasome core complex, alpha-subunit complex] GO:0031145 [anaphase-promoting complex-dependent proteasomal ubiquitin-dependent protein catabolic process] GO:0033209 [tumor necrosis factor-mediated signaling pathway] GO:0034641 [cellular nitrogen compound metabolic process] GO:0038061 [NIK/NF-kappaB signaling] GO:0038095 [Fc-epsilon receptor signaling pathway] GO:0042590 [antigen processing and presentation of exogenous peptide antigen via MHC class I] GO:0042981 [regulation of apoptotic process] GO:0043066 [negative regulation of apoptotic process] GO:0043161 [proteasome-mediated ubiquitin-dependent protein catabolic process] GO:0043488 [regulation of mRNA stability] GO:0044281 [small molecule metabolic process] GO:0045087 [innate immune response] GO:0048010 [vascular endothelial growth factor receptor signaling pathway] GO:0048011 [neurotrophin TRK receptor signaling pathway] GO:0050852 [T cell receptor signaling pathway] GO:0051436 [negative regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle] GO:0051437 [positive regulation of ubiquitin-protein ligase activity involved in regulation of mitotic cell cycle transition] GO:0051439 [regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle] GO:0051603 [proteolysis involved in cellular protein catabolic process] GO:0070062 [extracellular exosome] GO:0090090 [negative regulation of canonical Wnt signaling pathway] GO:0090263 [positive regulation of canonical Wnt signaling pathway]