We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
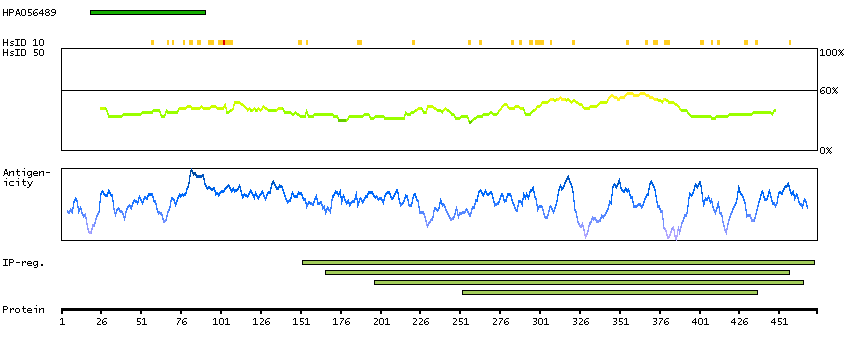
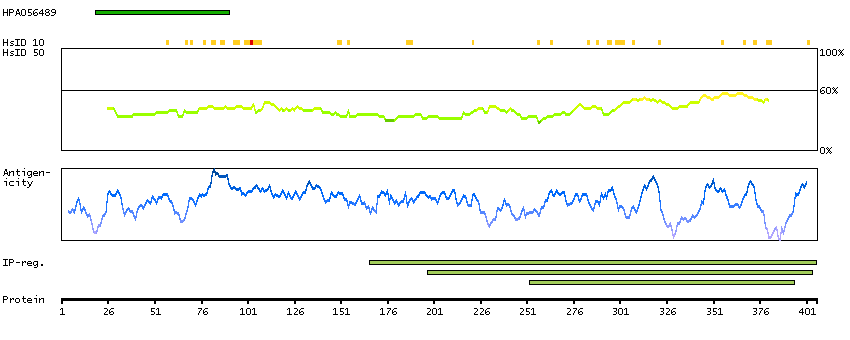
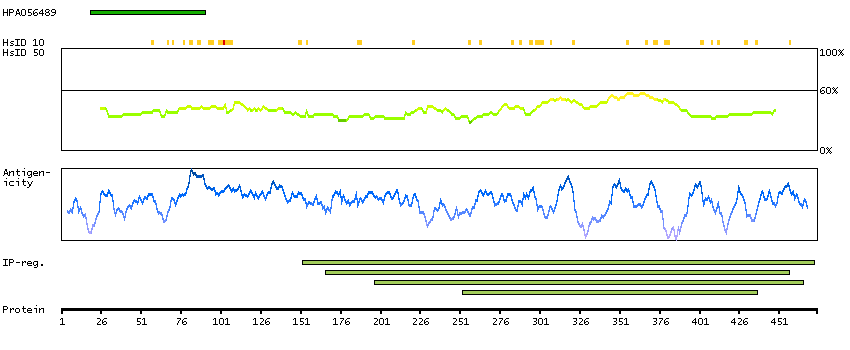
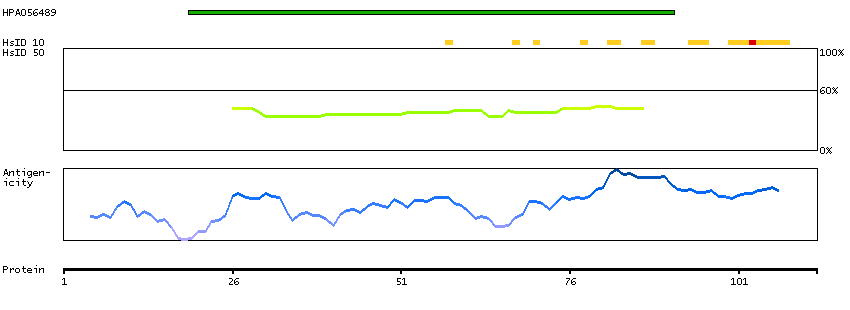
This gene encodes a member of the vaccinia-related kinase (VRK) family of serine/threonine protein kinases. In both human and mouse, this gene has substitutions at several residues within the ATP binding motifs that in other kinases have been shown to be required for catalysis. In vitro assays indicate the protein lacks phosphorylation activity. The protein, however, likely retains its substrate binding capability. This gene is widely expressed in human tissues and its protein localizes to the nucleus. Alternative splicing results in multiple transcripts encoding different isoforms. [provided by RefSeq, Jul 2008]
Enzymes Kinases CK1 Ser/Thr protein kinases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes Kinases CK1 Ser/Thr protein kinases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)