We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
This gene encodes a protein that homodimerizes and functions as a transcription factor which activates the expression of some key metabolic genes regulating cellular growth and nuclear genes required for respiration, heme biosynthesis, and mitochondrial DNA transcription and replication. The protein has also been associated with the regulation of neurite outgrowth. Alternative splicing results in multiple transcript variants. Confusion has occurred in bibliographic databases due to the shared symbol of NRF1 for this gene and for ""nuclear factor (erythroid-derived 2)-like 1"" which has an official symbol of NFE2L1. [provided by RefSeq, May 2014]
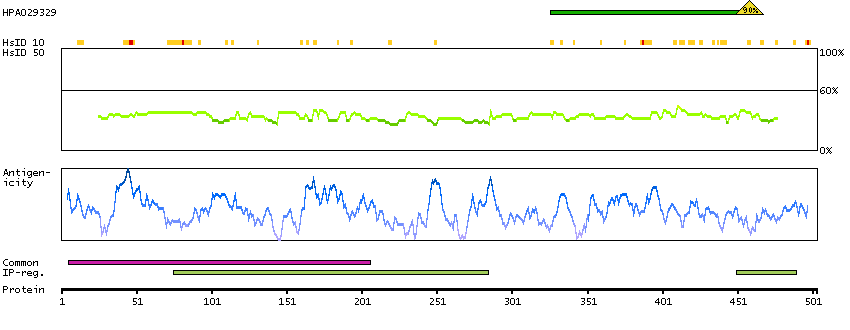
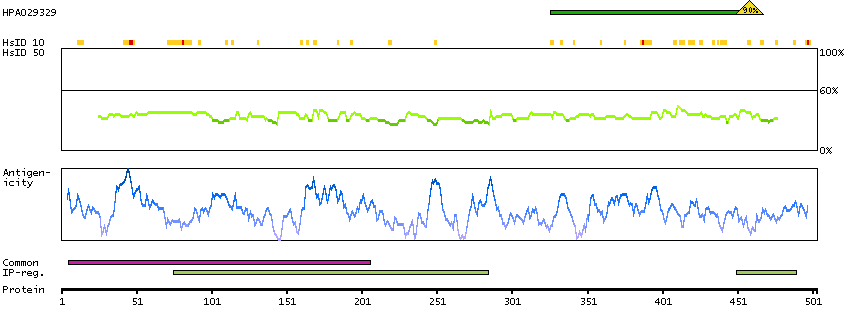
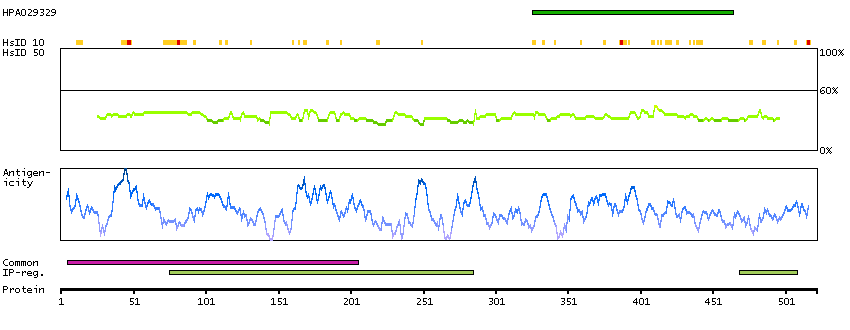
Predicted intracellular proteins Transcription factors Yet undefined DNA-binding domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000978 [RNA polymerase II core promoter proximal region sequence-specific DNA binding] GO:0001046 [core promoter sequence-specific DNA binding] GO:0001077 [transcriptional activator activity, RNA polymerase II core promoter proximal region sequence-specific binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0006091 [generation of precursor metabolites and energy] GO:0006357 [regulation of transcription from RNA polymerase II promoter] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006996 [organelle organization] GO:0007005 [mitochondrion organization] GO:0042803 [protein homodimerization activity] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0070062 [extracellular exosome]
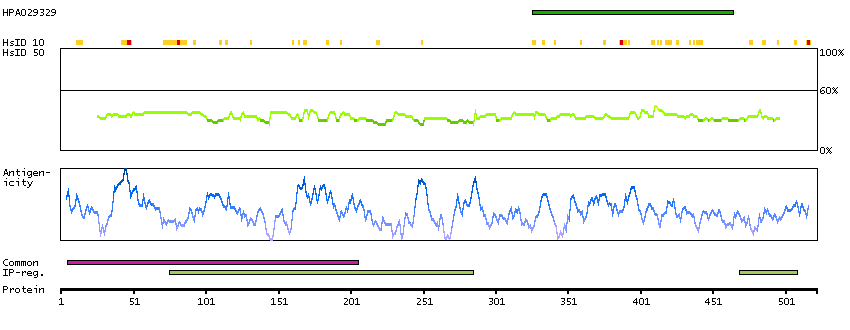
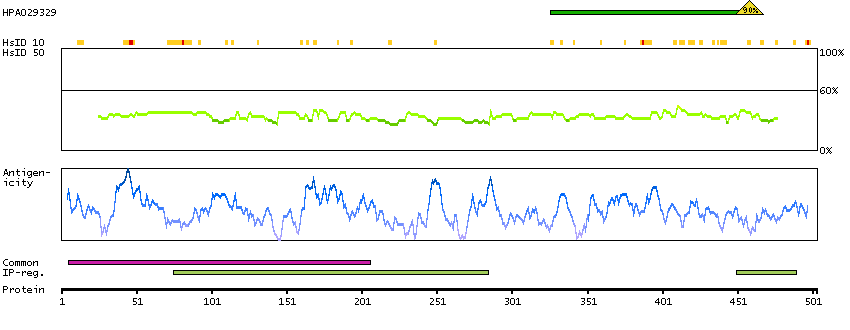
Predicted intracellular proteins Transcription factors Yet undefined DNA-binding domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000978 [RNA polymerase II core promoter proximal region sequence-specific DNA binding] GO:0001046 [core promoter sequence-specific DNA binding] GO:0001077 [transcriptional activator activity, RNA polymerase II core promoter proximal region sequence-specific binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0006091 [generation of precursor metabolites and energy] GO:0006357 [regulation of transcription from RNA polymerase II promoter] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006996 [organelle organization] GO:0007005 [mitochondrion organization] GO:0042803 [protein homodimerization activity] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0070062 [extracellular exosome]