We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
F-box and WD repeat domain containing 7, E3 ubiquitin protein ligase (HGNC Symbol)
Entrez gene summary
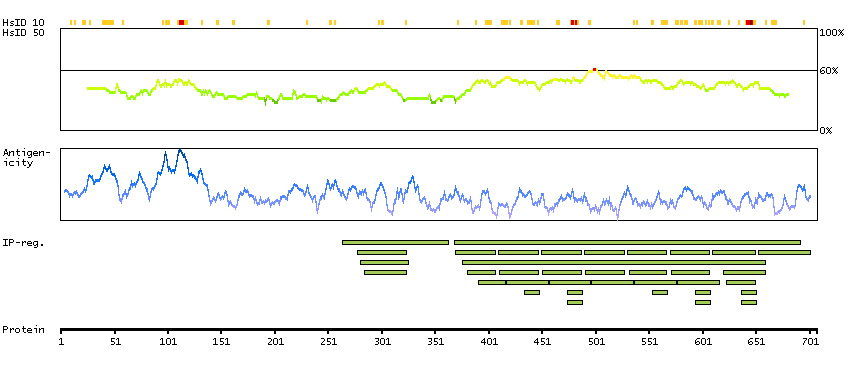
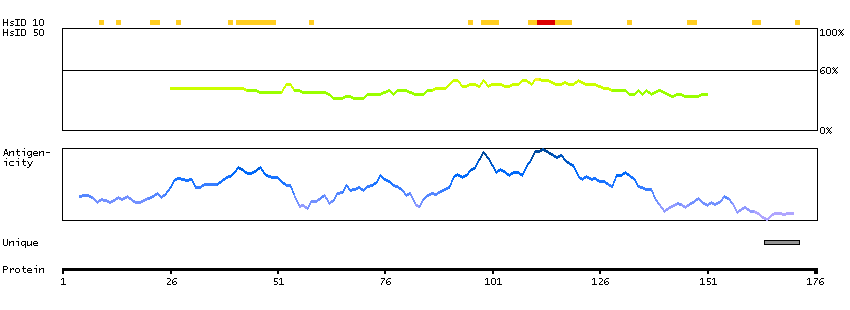
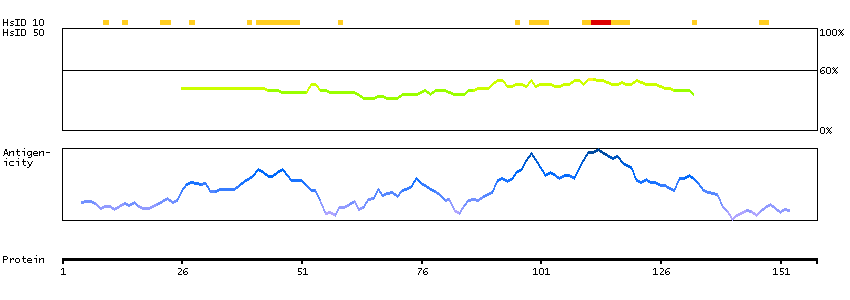
This gene encodes a member of the F-box protein family which is characterized by an approximately 40 amino acid motif, the F-box. The F-box proteins constitute one of the four subunits of ubiquitin protein ligase complex called SCFs (SKP1-cullin-F-box), which function in phosphorylation-dependent ubiquitination. The F-box proteins are divided into 3 classes: Fbws containing WD-40 domains, Fbls containing leucine-rich repeats, and Fbxs containing either different protein-protein interaction modules or no recognizable motifs. The protein encoded by this gene was previously referred to as FBX30, and belongs to the Fbws class; in addition to an F-box, this protein contains 7 tandem WD40 repeats. This protein binds directly to cyclin E and probably targets cyclin E for ubiquitin-mediated degradation. Mutations in this gene are detected in ovarian and breast cancer cell lines, implicating the gene's potential role in the pathogenesis of human cancers. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Mar 2012]