We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Cbl proto-oncogene, E3 ubiquitin protein ligase (HGNC Symbol)
Entrez gene summary
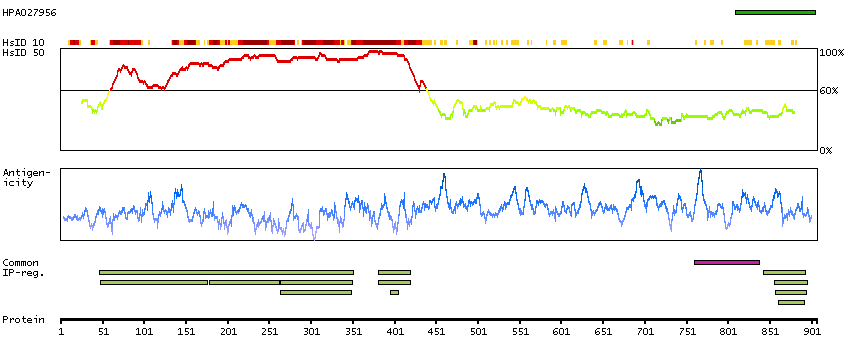
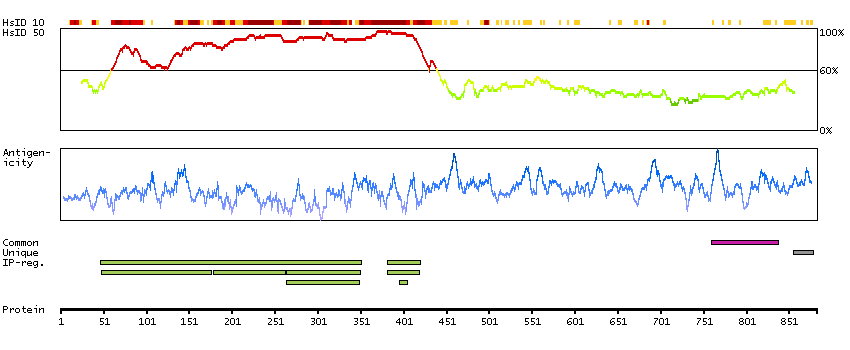
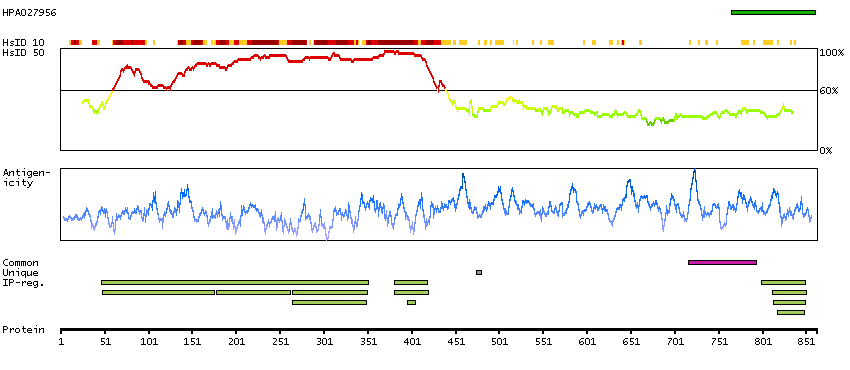
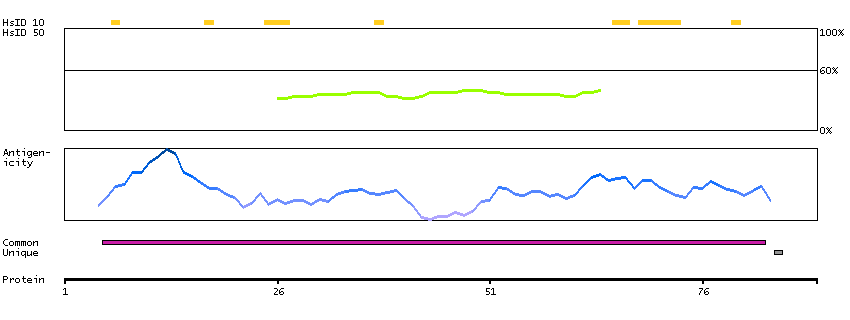
This gene is a proto-oncogene that encodes a RING finger E3 ubiquitin ligase. The encoded protein is one of the enzymes required for targeting substrates for degradation by the proteasome. This protein mediates the transfer of ubiquitin from ubiquitin conjugating enzymes (E2) to specific substrates. This protein also contains an N-terminal phosphotyrosine binding domain that allows it to interact with numerous tyrosine-phosphorylated substrates and target them for proteasome degradation. As such it functions as a negative regulator of many signal transduction pathways. This gene has been found to be mutated or translocated in many cancers including acute myeloid leukaemia. Mutations in this gene are also the cause of Noonan syndrome-like disorder. [provided by RefSeq, Mar 2012]
Predicted intracellular proteins Cancer-related genes Candidate cancer biomarkers COSMIC somatic mutations in cancer genes COSMIC Splicing Mutations COSMIC Somatic Mutations COSMIC Other Mutations COSMIC Missense Mutations COSMIC Translocations Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)