We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
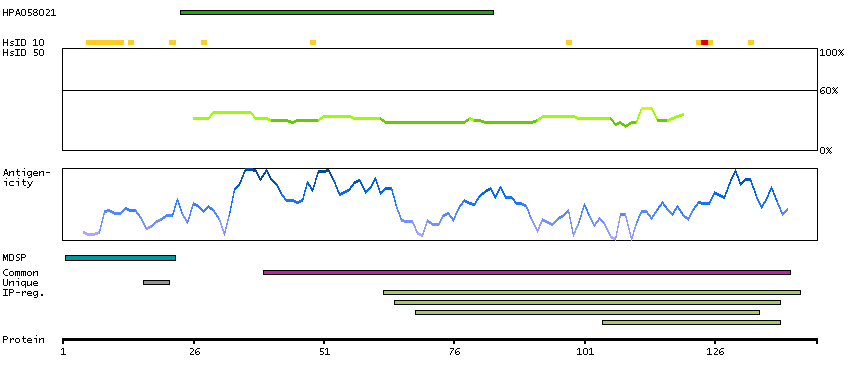
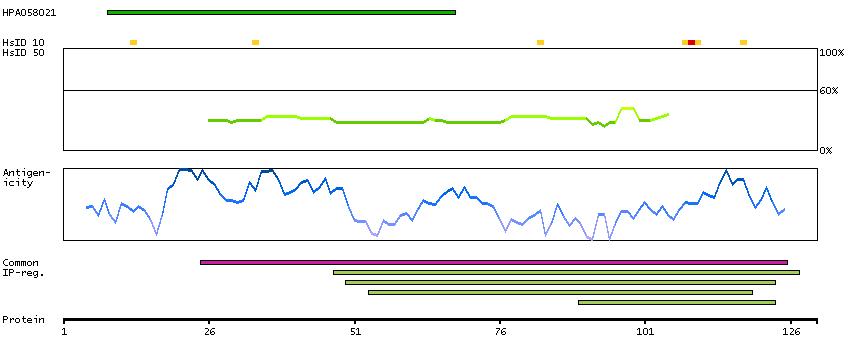
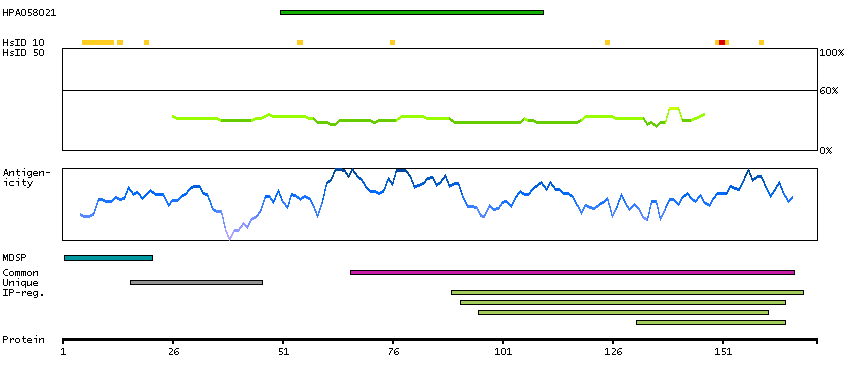
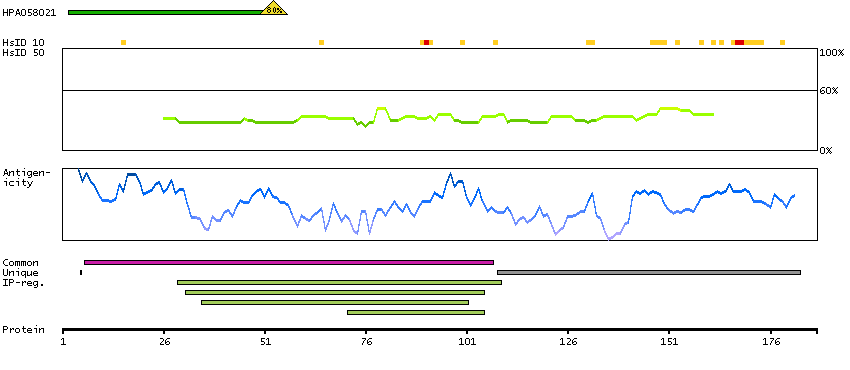
The product of this gene is a member of the saposin-like protein (SAPLIP) family and is located in the cytotoxic granules of T cells, which are released upon antigen stimulation. This protein is present in cytotoxic granules of cytotoxic T lymphocytes and natural killer cells, and it has antimicrobial activity against M. tuberculosis and other organisms. Alternatively spliced transcript variants encoding different isoforms have been identified. [provided by RefSeq, Jul 2008]
Transporters Transporter channels and pores Predicted secreted proteins Secreted proteins predicted by MDSEC SignalP predicted secreted proteins Phobius predicted secreted proteins SPOCTOPUS predicted secreted proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005615 [extracellular space] GO:0006968 [cellular defense response] GO:0031640 [killing of cells of other organism] GO:0042742 [defense response to bacterium] GO:0050832 [defense response to fungus]
Transporters Transporter channels and pores Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005615 [extracellular space] GO:0006968 [cellular defense response] GO:0031640 [killing of cells of other organism] GO:0042742 [defense response to bacterium] GO:0050832 [defense response to fungus]