We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
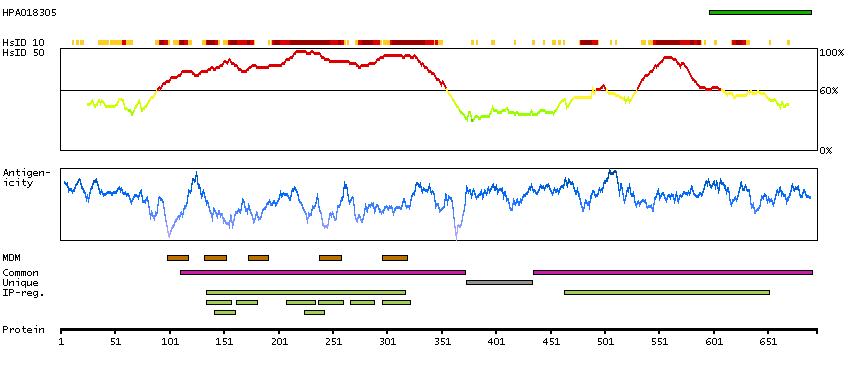
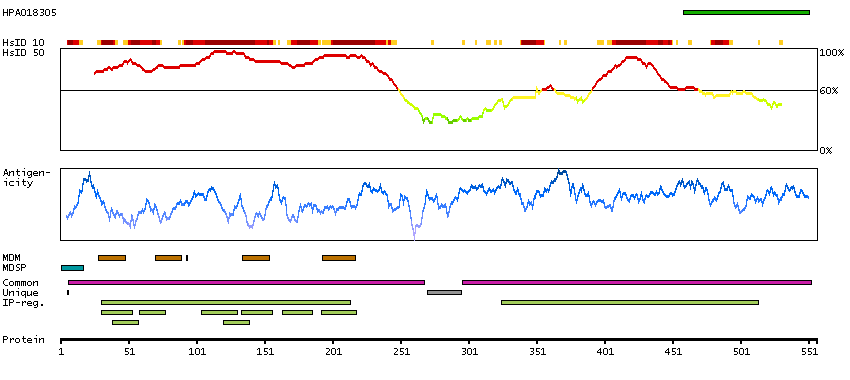
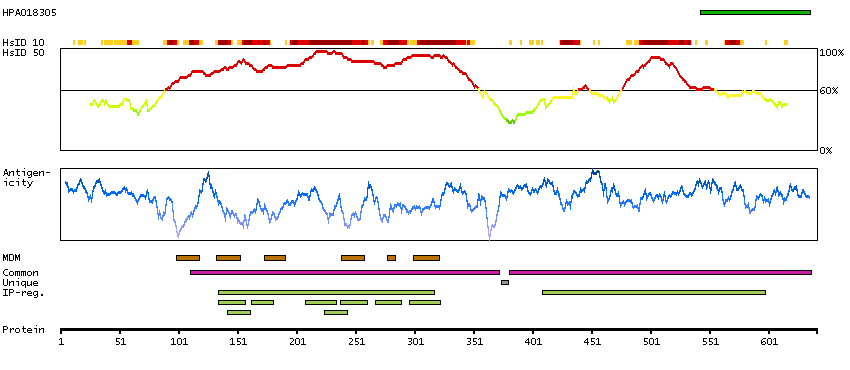
Potassium channel, voltage gated KQT-like subfamily Q, member 4 (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene forms a potassium channel that is thought to play a critical role in the regulation of neuronal excitability, particularly in sensory cells of the cochlea. The current generated by this channel is inhibited by M1 muscarinic acetylcholine receptors and activated by retigabine, a novel anti-convulsant drug. The encoded protein can form a homomultimeric potassium channel or possibly a heteromultimeric channel in association with the protein encoded by the KCNQ3 gene. Defects in this gene are a cause of nonsyndromic sensorineural deafness type 2 (DFNA2), an autosomal dominant form of progressive hearing loss. Two transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jul 2008]