We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
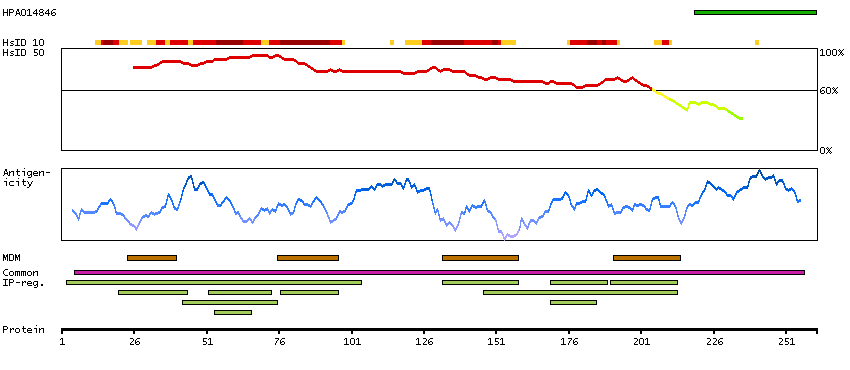
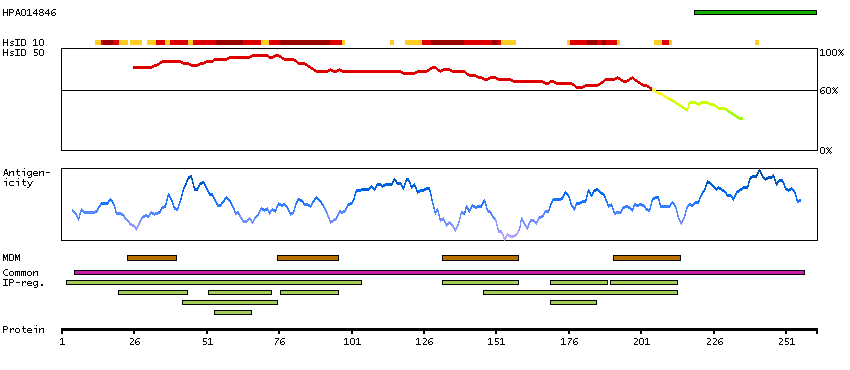
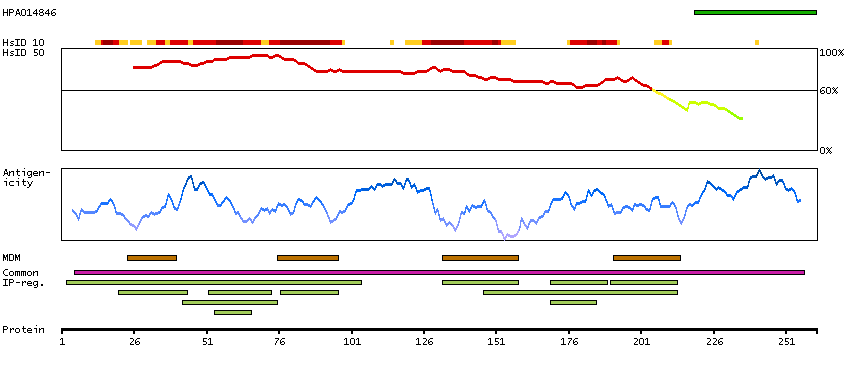
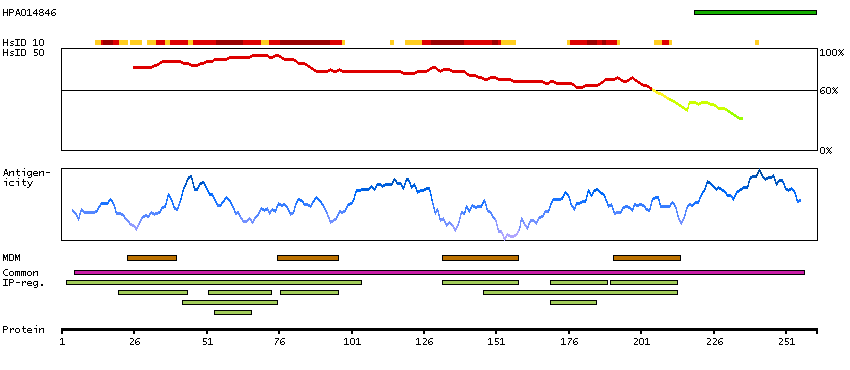
Gap junctions allow the transport of ions and metabolites between the cytoplasm of adjacent cells. They are formed by two hemichannels, made up of six connexin proteins assembled in groups. Each connexin protein has four transmembrane segments, two extracellular loops, a cytoplasmic loop formed between the two inner transmembrane segments, and the N- and C-terminus both being in the cytoplasm. The specificity of the gap junction is determined by which connexin proteins comprise the hemichannel. In the past, connexin protein names were based on their molecular weight, however the new nomenclature uses sequential numbers based on which form (alpha or beta) of the gap junction is present. This gene encodes one of the connexin proteins. Mutations in this gene have been found in some forms of deafness and in some families with hidrotic ectodermal dysplasia. [provided by RefSeq, Jul 2008]