We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
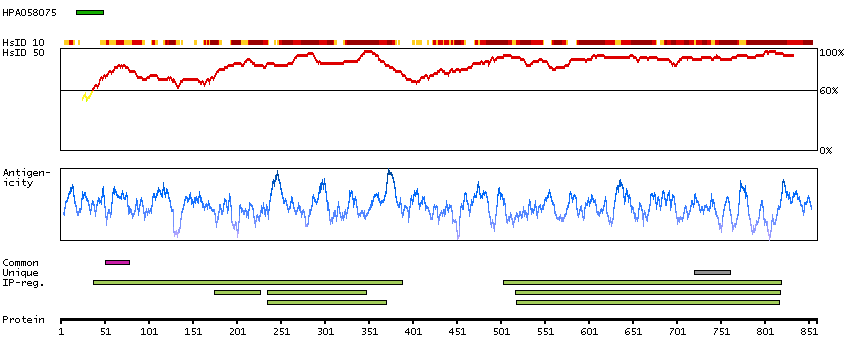
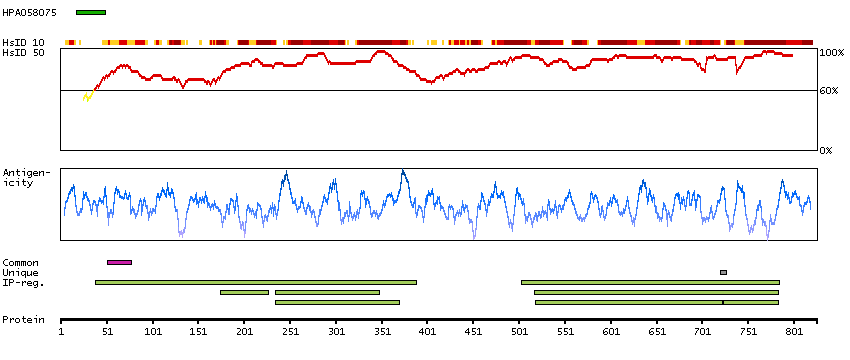
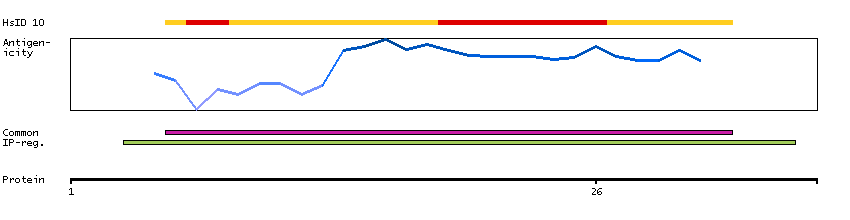
This gene encodes a member of the Argonaute family of proteins which play a role in RNA interference. The encoded protein is highly basic, and contains a PAZ domain and a PIWI domain. It may interact with dicer1 and play a role in short-interfering-RNA-mediated gene silencing. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Sep 2009]
Enzymes ENZYME proteins Hydrolases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Hydrolases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)