We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
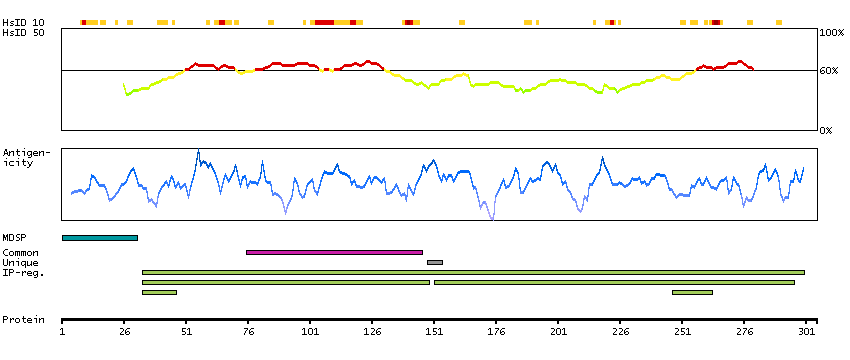
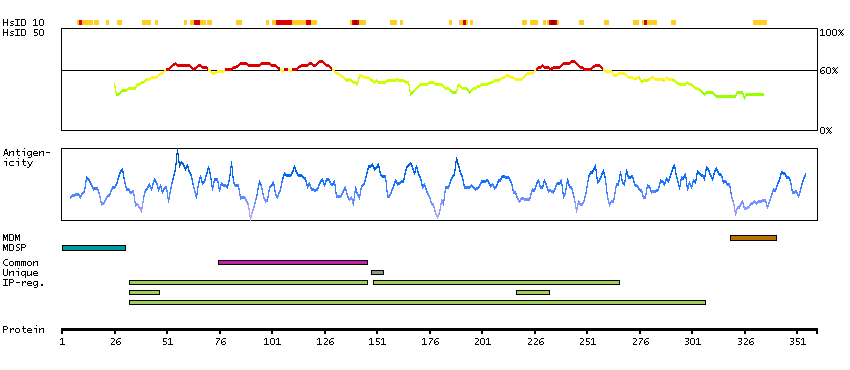
This gene encodes the beta subunit of lysosomal acid phosphatase (LAP). LAP is chemically and genetically distinct from red cell acid phosphatase. The encoded protein belongs to a family of distinct isoenzymes which hydrolyze orthophosphoric monoesters to alcohol and phosphate. LAP-deficiencies in mice cause multiple defects including bone structure alterations, lysosomal storage defects in the kidneys and central nervous system, and an increased tendency towards seizures. An enzymatically-inactive allele of LAP in mice exhibited a more severe phenotype than the null allele, and defects included cerebellum abnormalities, growth retardation, hair-follicle abnormalities, and an ataxia-like phenotype. Alternative splicing results in multiple transcript variants encoding different isoforms. [provided by RefSeq, Oct 2014]