We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
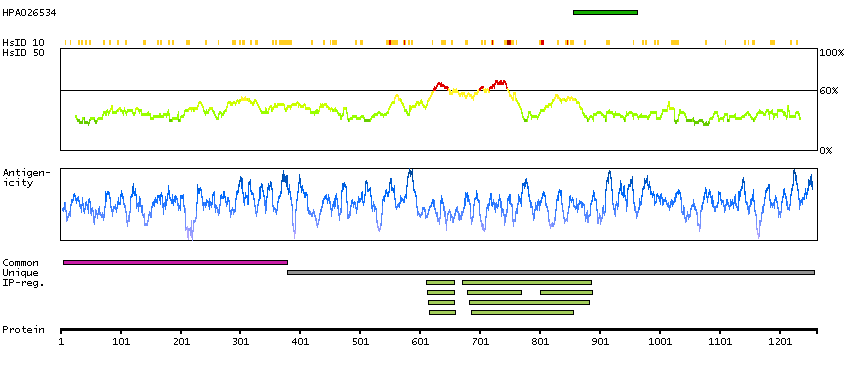
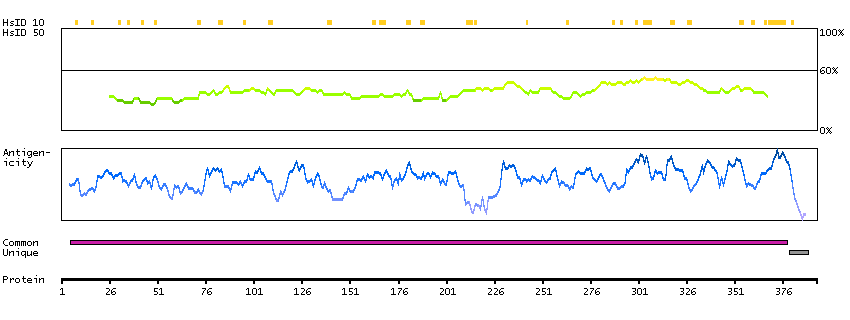
Q52LW3 [Direct mapping] Rho GTPase-activating protein 29
Show all
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005096 [GTPase activator activity] GO:0005829 [cytosol] GO:0007264 [small GTPase mediated signal transduction] GO:0007266 [Rho protein signal transduction] GO:0030165 [PDZ domain binding] GO:0043547 [positive regulation of GTPase activity] GO:0046872 [metal ion binding] GO:0051056 [regulation of small GTPase mediated signal transduction]