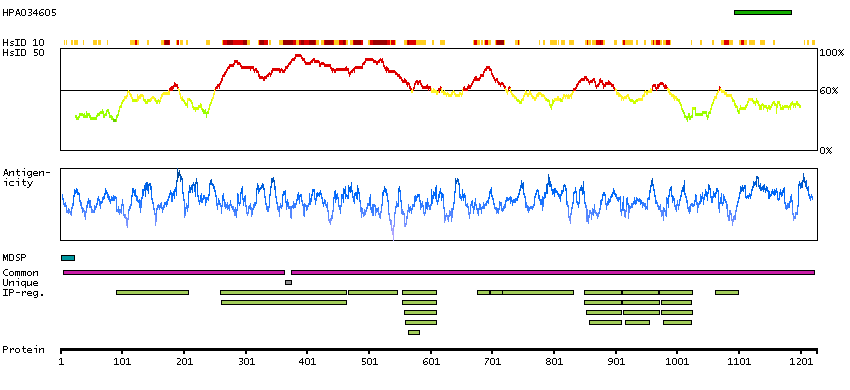
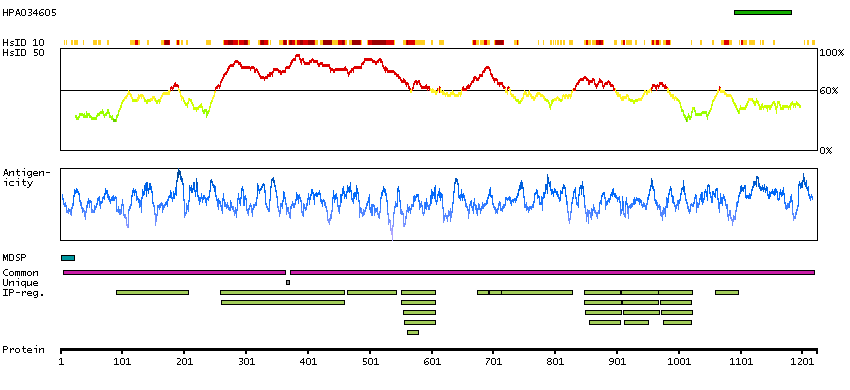
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
ADAM metallopeptidase with thrombospondin type 1 motif, 14 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motif) protein family. Members of the family share several distinct protein modules, including a propeptide region, a metalloproteinase domain, a disintegrin-like domain, and a thrombospondin type 1 (TS) motif. Individual members of this family differ in the number of C-terminal TS motifs, and some have unique C-terminal domains. This gene is highly similar to two family members, ADAMTS2 and ADAMTS3, in its sequence and gene structure, and the encoded protein shares the aminoprocollagen peptidase activity with the protein products encoded by ADAMTS2 and ADAMTS3. Various transcript variants of this gene have been identified. They result from the use of two different promoters and transcription initiation sites as well as alternative splicing sites. The full length nature of some transcripts has not been defined. [provided by RefSeq, Jul 2008]