We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Inherited mutations in BRCA1 and this gene, BRCA2, confer increased lifetime risk of developing breast or ovarian cancer. Both BRCA1 and BRCA2 are involved in maintenance of genome stability, specifically the homologous recombination pathway for double-strand DNA repair. The BRCA2 protein contains several copies of a 70 aa motif called the BRC motif, and these motifs mediate binding to the RAD51 recombinase which functions in DNA repair. BRCA2 is considered a tumor suppressor gene, as tumors with BRCA2 mutations generally exhibit loss of heterozygosity (LOH) of the wild-type allele. [provided by RefSeq, Dec 2008]
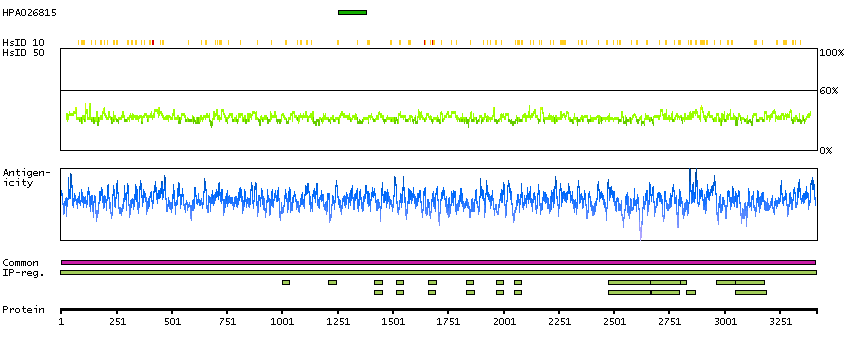
P51587 [Direct mapping] Breast cancer type 2 susceptibility protein
Show all
Predicted intracellular proteins Cancer-related genes Candidate cancer biomarkers Mutational cancer driver genes COSMIC somatic mutations in cancer genes COSMIC Splicing Mutations COSMIC Somatic Mutations COSMIC Nonsense Mutations COSMIC Missense Mutations COSMIC Large Deletions COSMIC Germline Mutations COSMIC Frameshift Mutations Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)