We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Fractions of cells showed weak nuclear and/or cytoplasmic expression.
DATA RELIABILITY
Data reliability description
Antibody staining mainly consistent with RNA expression data. Tissue location of RNA and protein might differ due to short half-life of wild-type p53 protein.
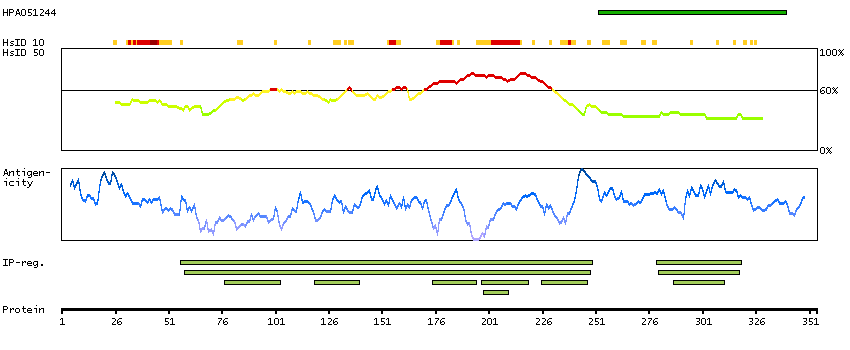
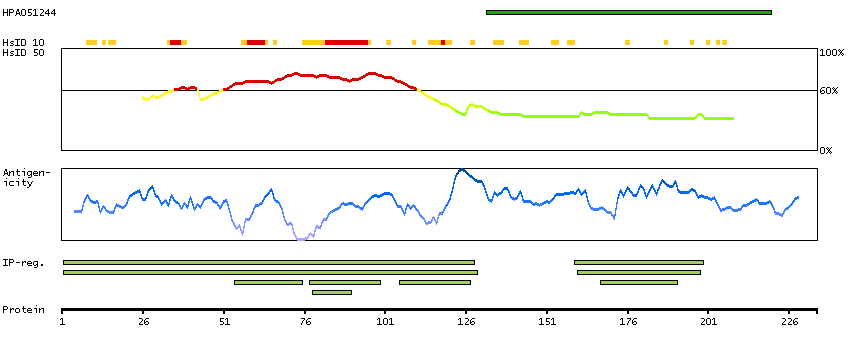
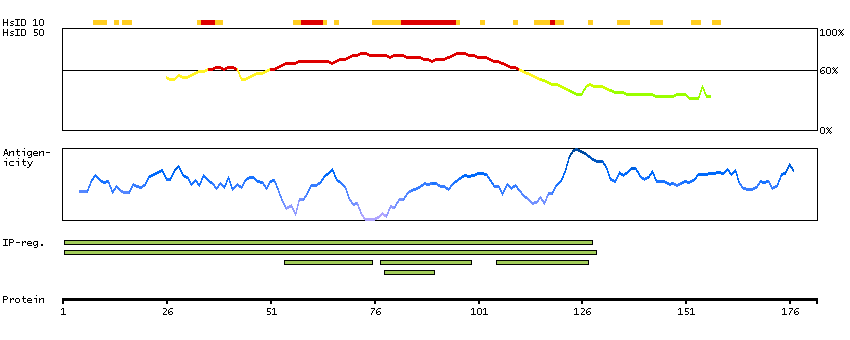
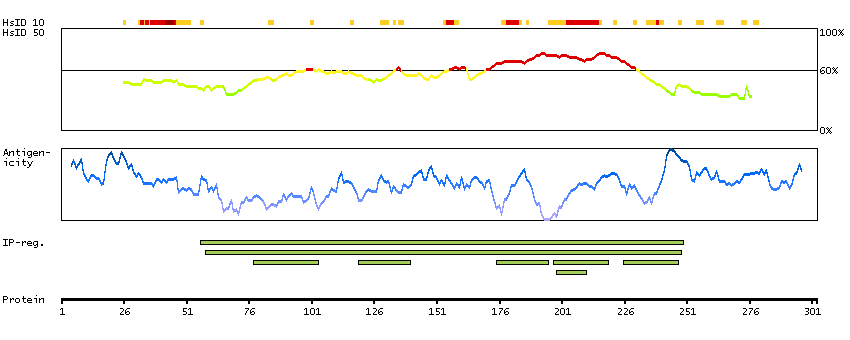
This gene encodes a tumor suppressor protein containing transcriptional activation, DNA binding, and oligomerization domains. The encoded protein responds to diverse cellular stresses to regulate expression of target genes, thereby inducing cell cycle arrest, apoptosis, senescence, DNA repair, or changes in metabolism. Mutations in this gene are associated with a variety of human cancers, including hereditary cancers such as Li-Fraumeni syndrome. Alternative splicing of this gene and the use of alternate promoters result in multiple transcript variants and isoforms. Additional isoforms have also been shown to result from the use of alternate translation initiation codons (PMIDs: 12032546, 20937277). [provided by RefSeq, Feb 2013]