We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
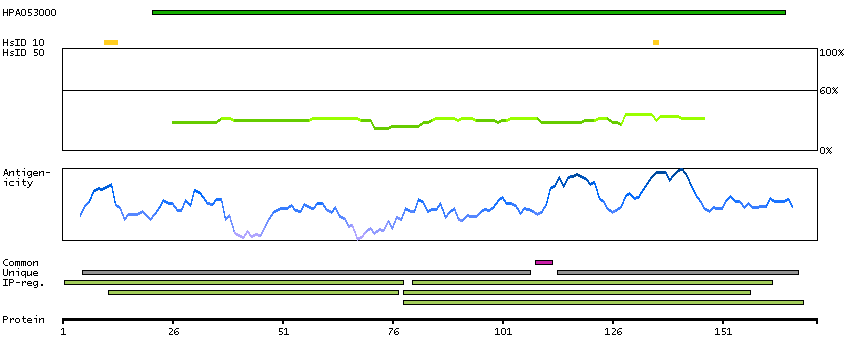
Polymerase (RNA) II (DNA directed) polypeptide G (HGNC Symbol)
Entrez gene summary
This gene encodes the seventh largest subunit of RNA polymerase II, the polymerase responsible for synthesizing messenger RNA in eukaryotes. The protein functions in transcription initiation, and is also thought to help stabilize transcribing polyermase molecules during elongation. [provided by RefSeq, Jan 2009]