We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
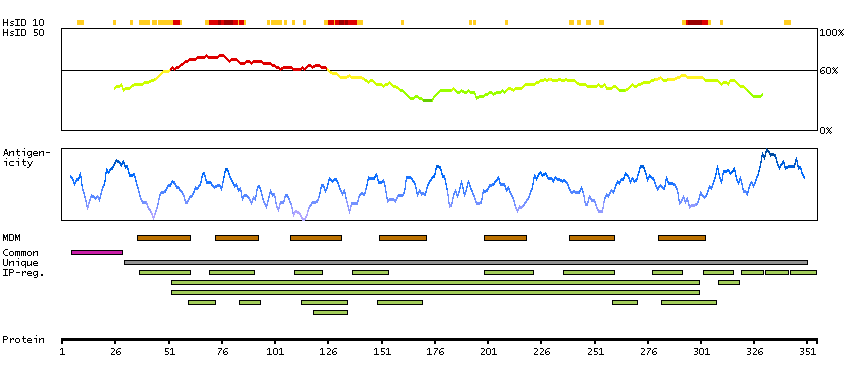
This gene encodes a member of the beta chemokine receptor family, which is predicted to be a seven transmembrane protein similar to G protein-coupled receptors. Chemokines and their receptors are important for the migration of various cell types into the inflammatory sites. This receptor protein preferentially expresses in the thymus. I-309, thymus activation-regulated cytokine (TARC) and macrophage inflammatory protein-1 beta (MIP-1 beta) have been identified as ligands of this receptor. Studies of this receptor and its ligands suggested its role in regulation of monocyte chemotaxis and thymic cell apoptosis. More specifically, this receptor may contribute to the proper positioning of activated T cells within the antigenic challenge sites and specialized areas of lymphoid tissues. This gene is located at the chemokine receptor gene cluster region. [provided by RefSeq, Jul 2008]