We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
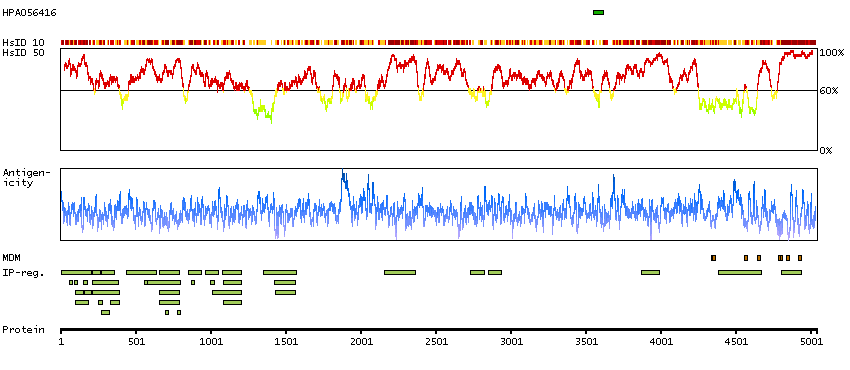
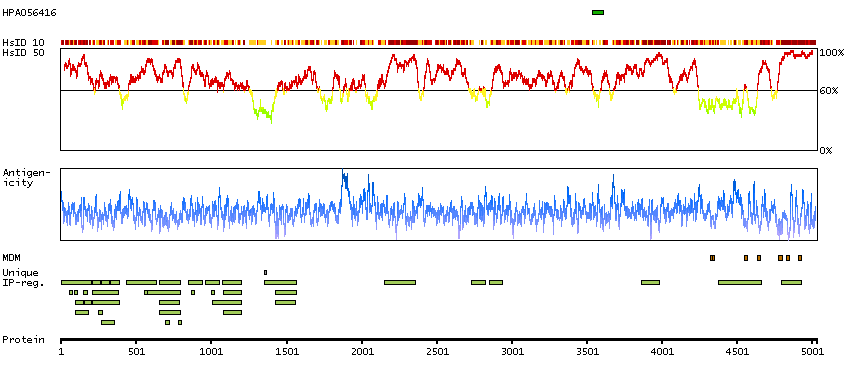
This gene encodes a ryanodine receptor found in skeletal muscle. The encoded protein functions as a calcium release channel in the sarcoplasmic reticulum but also serves to connect the sarcoplasmic reticulum and transverse tubule. Mutations in this gene are associated with malignant hyperthermia susceptibility, central core disease, and minicore myopathy with external ophthalmoplegia. Alternatively spliced transcripts encoding different isoforms have been described. [provided by RefSeq, Jul 2008]