We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
This gene product appears to function as a negative regulator of the ras signal transduction pathway. Mutations in this gene have been linked to neurofibromatosis type 1, juvenile myelomonocytic leukemia and Watson syndrome. The mRNA for this gene is subject to RNA editing (CGA>UGA->Arg1306Term) resulting in premature translation termination. Alternatively spliced transcript variants encoding different isoforms have also been described for this gene. [provided by RefSeq, Jul 2008]
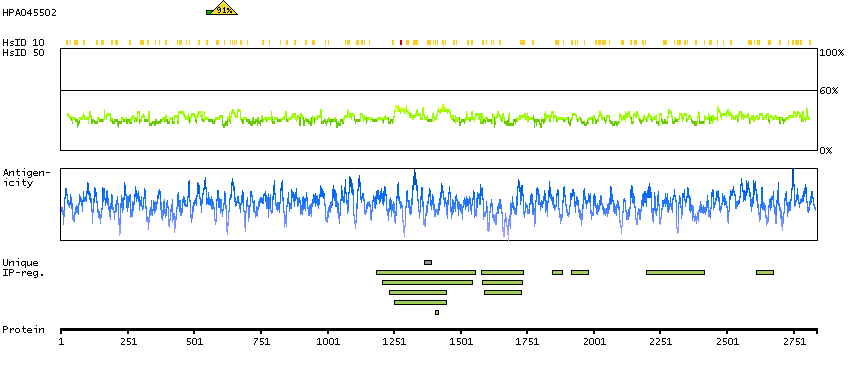
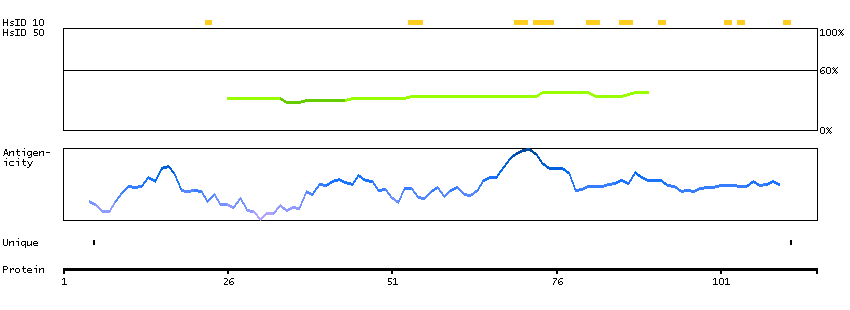
MEMSAT-SVM predicted membrane proteins SPOCTOPUS predicted membrane proteins THUMBUP predicted membrane proteins Predicted intracellular proteins RAS pathway related proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes COSMIC somatic mutations in cancer genes COSMIC Splicing Mutations COSMIC Somatic Mutations COSMIC Other Mutations COSMIC Nonsense Mutations COSMIC Missense Mutations COSMIC Large Deletions COSMIC Germline Mutations COSMIC Frameshift Mutations Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005488 [binding] GO:0005737 [cytoplasm] GO:0007165 [signal transduction] GO:0031235 [intrinsic component of the cytoplasmic side of the plasma membrane] GO:0043087 [regulation of GTPase activity] GO:0046580 [negative regulation of Ras protein signal transduction]
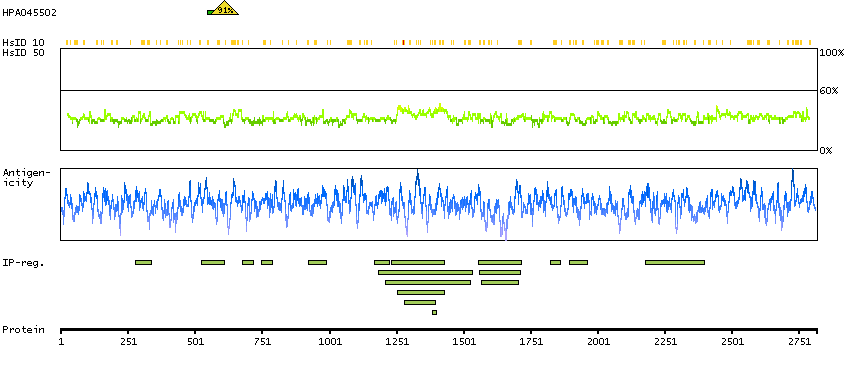
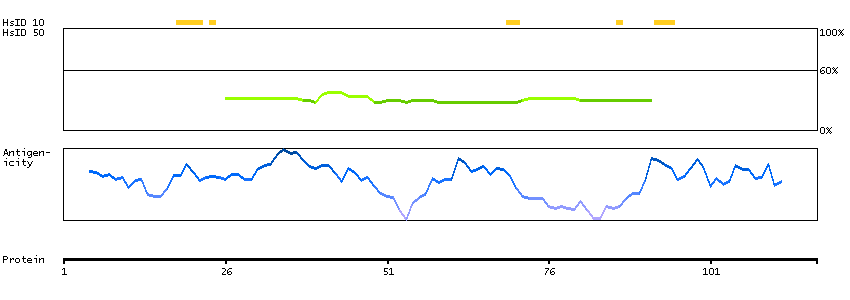
Predicted intracellular proteins RAS pathway related proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes COSMIC somatic mutations in cancer genes COSMIC Splicing Mutations COSMIC Somatic Mutations COSMIC Other Mutations COSMIC Nonsense Mutations COSMIC Missense Mutations COSMIC Large Deletions COSMIC Germline Mutations COSMIC Frameshift Mutations Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005737 [cytoplasm] GO:0031235 [intrinsic component of the cytoplasmic side of the plasma membrane] GO:0046580 [negative regulation of Ras protein signal transduction]
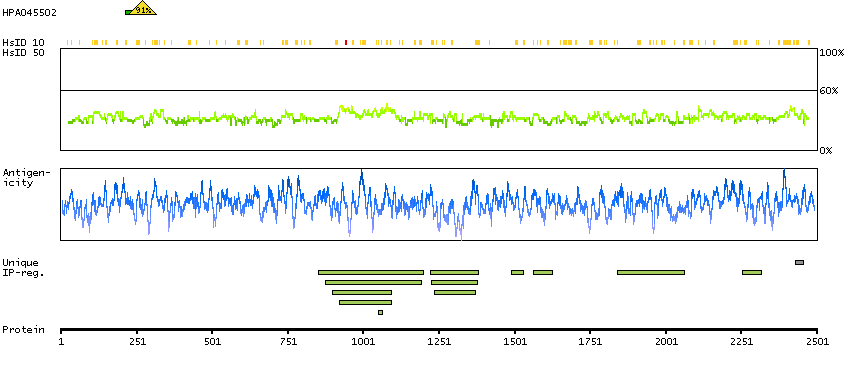
Predicted intracellular proteins RAS pathway related proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes COSMIC somatic mutations in cancer genes COSMIC Splicing Mutations COSMIC Somatic Mutations COSMIC Other Mutations COSMIC Nonsense Mutations COSMIC Missense Mutations COSMIC Large Deletions COSMIC Germline Mutations COSMIC Frameshift Mutations Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005737 [cytoplasm] GO:0031235 [intrinsic component of the cytoplasmic side of the plasma membrane] GO:0046580 [negative regulation of Ras protein signal transduction]