We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Sodium channel, voltage gated, type VIII alpha subunit (HGNC Symbol)
Entrez gene summary
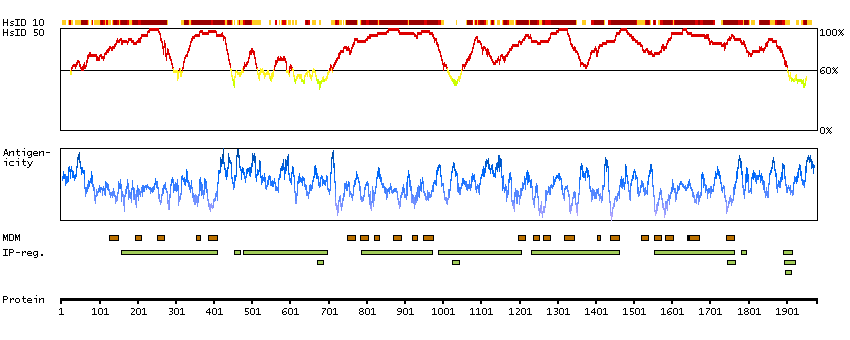
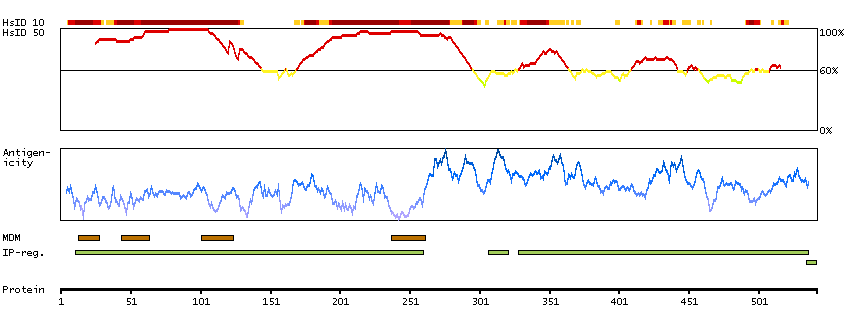
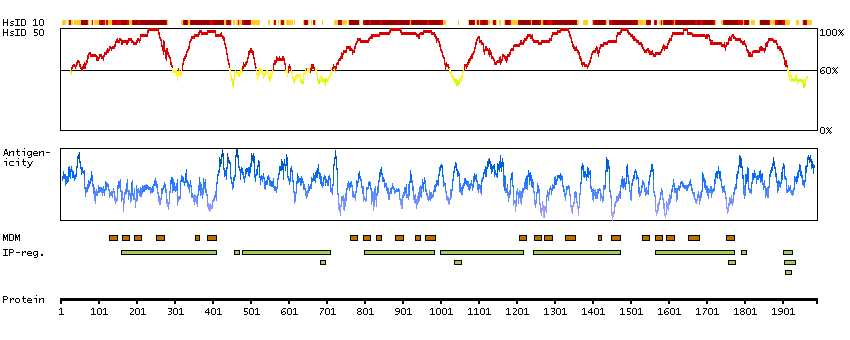
This gene encodes a member of the sodium channel alpha subunit gene family. The encoded protein forms the ion pore region of the voltage-gated sodium channel. This protein is essential for the rapid membrane depolarization that occurs during the formation of the action potential in excitable neurons. Mutations in this gene are associated with mental retardation, pancerebellar atrophy and ataxia. Alternate splicing results in multiple transcript variants.[provided by RefSeq, May 2010]