We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
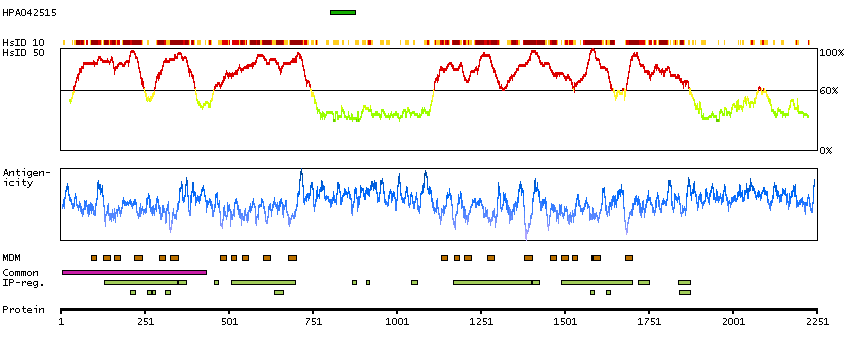
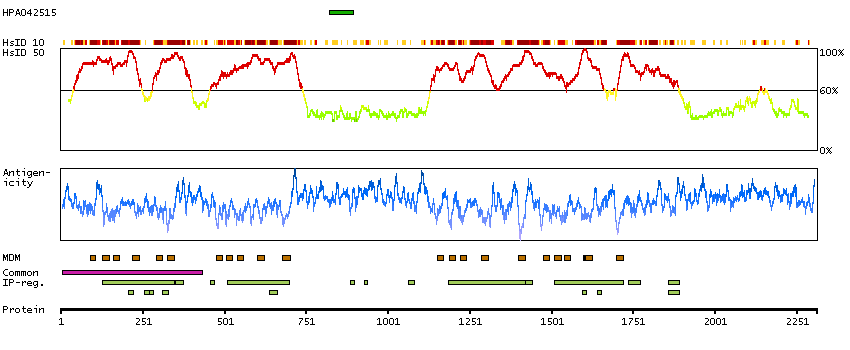
Calcium channel, voltage-dependent, R type, alpha 1E subunit (HGNC Symbol)
Entrez gene summary
Voltage-dependent calcium channels are multisubunit complexes consisting of alpha-1, alpha-2, beta, and delta subunits in a 1:1:1:1 ratio. These channels mediate the entry of calcium ions into excitable cells, and are also involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release, gene expression, cell motility, cell division and cell death. This gene encodes the alpha-1E subunit of the R-type calcium channels, which belong to the 'high-voltage activated' group that maybe involved in the modulation of firing patterns of neurons important for information processing. Alternatively spliced transcript variants encoding different isoforms have been described for this gene. [provided by RefSeq, Apr 2011]